Ruoli multidimensionali di corpi chetonici
I corpi chetonici sono creati dal fegato e utilizzati come fonte di energia quando il glucosio non è prontamente disponibile nel corpo umano. I due corpi chetonici principali sono l'acetoacetato (AcAc) e il 3-beta-idrossibutirrato (3HB), mentre l'acetone è il terzo e meno abbondante corpo chetonico. I chetoni sono sempre presenti nel sangue e il loro livello aumenta durante il digiuno e l'esercizio prolungatochetogenesi è il processo biochimico mediante il quale gli organismi producono corpi chetonici attraverso la scissione di acidi grassi e amminoacidi chetogenici.
I corpi chetonici sono generati principalmente nel mitocondri di cellule del fegato. La chetogenesi si verifica quando vi sono bassi livelli di glucosio nel sangue, in particolare dopo che sono state esaurite altre riserve di carboidrati cellulari, come il glicogeno. Questo meccanismo può anche verificarsi quando non vi sono quantità sufficienti di insulina. La produzione di corpi chetonici viene infine avviata per rendere disponibile l'energia immagazzinata nel corpo umano sotto forma di acidi grassi. La chetogenesi si verifica nei mitocondri dove è regolata indipendentemente.
Astratto
Il metabolismo del corpo chetone è un nodo centrale nell'omeostasi fisiologica. In questa recensione, discutiamo di come i chetoni servono ruoli metabolici discreti a regolazione fine che ottimizzano le prestazioni di organi e organismi in vari resti di nutrienti e proteggono da infiammazioni e lesioni in sistemi di organi multipli. Tradizionalmente visti come substrati metabolici arruolati solo nella restrizione dei carboidrati, recenti osservazioni sottolineano l'importanza dei corpi chetonici come metabolici metabolici e di segnalazione vitali quando i carboidrati sono abbondanti. A complemento di un repertorio di opzioni terapeutiche conosciute per le malattie del sistema nervoso, sono emersi i ruoli potenziali per i corpi chetonici nel cancro, così come i ruoli protettivi intriganti nel cuore e nel fegato, aprendo le opzioni terapeutiche nelle malattie legate all'obesità e cardiovascolari. Le controversie nel metabolismo e segnalazione dei chetoni sono discusse per conciliare il dogma classico con le osservazioni contemporanee.
Introduzione
I corpi chetonici sono una fonte vitale alternativa di combustibile metabolico per tutti i domini della vita, eucarya, batteri e archei (Aneja et al., 2002; Cahill GF Jr, 2006; Krishnakumar et al., 2008). Il metabolismo del corpo chetonico negli esseri umani è stato sfruttato per alimentare il cervello durante periodi episodici di privazione dei nutrienti. I corpi chetonici sono intrecciati con le vie metaboliche cruciali dei mammiferi come l'ossidazione β (FAO), il ciclo degli acidi tricarbossilici (TCA), la gluconeogenesi, la lipogenesi de novo (DNL) e la biosintesi degli steroli. Nei mammiferi, i corpi chetonici sono prodotti prevalentemente nel fegato dall'acetil-CoA derivato dalla FAO e vengono trasportati nei tessuti extraepatici per l'ossidazione terminale. Questa fisiologia fornisce un carburante alternativo che è aumentato da periodi relativamente brevi di digiuno, che aumenta la disponibilità di acidi grassi e diminuisce la disponibilità di carboidrati (Cahill GF Jr, 2006; McGarry e Foster, 1980; Robinson e Williamson, 1980). L'ossidazione del corpo chetonico diventa un contributo significativo al metabolismo energetico complessivo dei mammiferi all'interno dei tessuti extraepatici in una miriade di stati fisiologici, tra cui digiuno, fame, periodo neonatale, post-esercizio, gravidanza e aderenza a diete a basso contenuto di carboidrati. Le concentrazioni corporee di chetoni totali circolanti in esseri umani adulti sani normalmente mostrano oscillazioni circadiane tra circa 100-250 M, salgono a ~ 1 mM dopo un esercizio prolungato o 24 ore di digiuno e possono accumularsi fino a 20 mM in stati patologici come la chetoacidosi diabetica ( Cahill GF Jr, 2006; Johnson et al., 1969b; Koeslag et al., 1980; Robinson e Williamson, 1980; Wildenhoff et al., 1974). Il fegato umano produce fino a 300 g di corpi chetonici al giorno (Balasse e Fery, 1989), che contribuiscono tra il 5 e il 20% del dispendio energetico totale negli stati nutriti, a digiuno e affamati (Balasse et al., 1978; Cox et al. al., 2016).
Recenti studi ora evidenziano ruoli imperativi per corpi chetonici nel metabolismo delle cellule dei mammiferi, omeostasi e segnalazione in un'ampia varietà di stati fisiologici e patologici. Oltre a servire come combustibile energetico per i tessuti extraepatici come cervello, cuore o muscolo scheletrico, i corpi chetonici svolgono ruoli chiave come mediatori segnalatori, fattori di modificazione della proteina post-traduzionale (PTM) e modulatori dell'infiammazione e dello stress ossidativo. In questa recensione, forniamo sia visioni classiche che moderne dei ruoli pleiotropici dei corpi chetonici e del loro metabolismo.
Panoramica del metabolismo del corpo chetone
Il tasso di chetogenesi epatica è regolato da una serie orchestrata di trasformazioni fisiologiche e biochimiche del grasso. I regolatori primari includono la lipolisi degli acidi grassi dai triacilgliceroli, il trasporto verso e attraverso la membrana plasmatica degli epatociti, il trasporto nei mitocondri tramite la carnitina palmitoiltransferasi 1 (CPT1), la spirale di α-ossidazione, l'attività del ciclo TCA e le concentrazioni intermedie, il potenziale redox e i regolatori ormonali di questi processi, prevalentemente glucagone e insulina [recensito in (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al., 1983; Kahn et al., 2005; McGarry e Foster , 1980; Williamson et al., 1969)]. Classicamente la chetogenesi è vista come una via di ricaduta, in cui l'acetil-CoA derivato dall'ossidazione α supera l'attività di citrato sintasi e / o la disponibilità di ossalacetato per la condensazione per formare citrato. Gli intermedi a tre atomi di carbonio mostrano attività anti-chetogenica, presumibilmente a causa della loro capacità di espandere il pool di ossalacetato per il consumo di acetil-CoA, ma la concentrazione di acetil-CoA epatica da sola non determina la velocità chetogenica (Foster, 1967; Rawat e Menahan, 1975; Williamson et al., 1969). La regolazione della chetogenesi da eventi ormonali, trascrizionali e post-traduzionali insieme supportano l'idea che i meccanismi molecolari che regolano la velocità chetogenica rimangono non completamente compresi (vedi Regolamento di HMGCS2 e SCOT / OXCT1).
La chetogenesi si verifica principalmente nella matrice mitocondriale epatica a velocità proporzionali all'ossidazione totale dei grassi. Dopo il trasporto delle catene aciliche attraverso le membrane mitocondriali e l'ossidazione a, l'isoforma mitocondriale della 3-idrossimetilglutaril-CoA sintasi (HMGCS2) catalizza il destino commettendo la condensazione dell'acetoacetil-CoA (AcAc-CoA) e dell'acetil-CoA per generare HMGCS1 (Fig. 1A). L'HMG-CoA liasi (HMGCL) scinde l'HMG-CoA per liberare acetil-CoA e acetoacetato (AcAc), e quest'ultimo viene ridotto a d -? - idrossibutirrato (d-? OHB) dalla d-? OHB deidrogenasi mitocondriale dipendente dalla fosfatidilcolina ( BDH1975) in una reazione di quasi equilibrio accoppiata a NAD + / NADH (Bock e Fleischer, 1960; LEHNINGER et al., 1). La costante di equilibrio BDH1 favorisce la produzione di d-? OHB, ma il rapporto tra corpi chetonici AcAc / d-? OHB è direttamente proporzionale al rapporto NAD + / NADH mitocondriale, e quindi l'attività ossidoreduttasi BDH1969 modula il potenziale redox mitocondriale (Krebs et al., 1967; Williamson et al., 1929). AcAc può anche decarbossilare spontaneamente in acetone (Pedersen, 7), la fonte di odore dolce negli esseri umani che soffrono di chetoacidosi (cioè, corpi chetonici sierici totali> ~ 3.6 mM; AcAc pKa 4.7,? OHB pKa 1). I meccanismi attraverso i quali i corpi chetonici vengono trasportati attraverso la membrana interna mitocondriale non sono noti, ma AcAc / d-? OHB vengono rilasciati dalle cellule tramite trasportatori monocarbossilati (nei mammiferi, MCT 2 e 16, noti anche come vettori di soluti 1A membri della famiglia 7 e 2011) e trasportati in circolo ai tessuti extraepatici per l'ossidazione terminale (Cotter et al., 2012; Halestrap e Wilson, 2012; Halestrap, 2012; Hugo et al., 1940). Le concentrazioni dei corpi chetonici circolanti sono più alte di quelle nei tessuti extraepatici (Harrison e Long, 1) indicando che i corpi chetonici sono trasportati lungo un gradiente di concentrazione. Le mutazioni con perdita di funzione in MCTXNUMX sono associate ad attacchi spontanei di chetoacidosi, suggerendo un ruolo critico nell'importazione del corpo chetonico.

Ad eccezione della potenziale diversione dei corpi chetonici in destini non ossidativi (vedere Destini metabolici non ossidativi dei corpi chetonici), gli epatociti non hanno la capacità di metabolizzare i corpi chetonici che producono. I corpi chetonici sintetizzati de novo dal fegato sono (i) catabolizzati nei mitocondri dei tessuti extraepatici in acetil-CoA, che è disponibile per il ciclo TCA per l'ossidazione terminale (Fig. 1A), (ii) deviato alle vie di lipogenesi o sintesi degli steroli ( Fig. 1B) o (iii) escreto nelle urine. Come combustibile energetico alternativo, i corpi chetonici sono ossidati avidamente nel cuore, nei muscoli scheletrici e nel cervello (Balasse e Fery, 1989; Bentourkia et al., 2009; Owen et al., 1967; Reichard et al., 1974; Sultan, 1988; ). Il BDH1 mitocondriale extraepatico catalizza la prima reazione di ossidazione? OHB, convertendola in AcAc di ritorno (LEHNINGER et al., 1960; Sandermann et al., 1986). Una d-? OHB-deidrogenasi citoplasmatica (BDH2) con solo il 20% di identità di sequenza per BDH1 ha un elevato Km per i corpi chetonici e svolge anche un ruolo nell'omeostasi del ferro (Davuluri et al., 2016; Guo et al., 2006) . Nella matrice mitocondriale extraepatica, AcAc viene attivato in AcAc-CoA attraverso lo scambio di una frazione CoA da succinil-CoA in una reazione catalizzata da un'unica transferasi CoA di mammifero, succinil-CoA: 3-ossoacido-CoA transferasi (SCOT, CoA transferasi; codificato da OXCT1), attraverso una reazione di quasi equilibrio. L'energia libera rilasciata dall'idrolisi di AcAc-CoA è maggiore di quella del succinil-CoA, favorendo la formazione di AcAc. Pertanto, il flusso ossidativo del corpo chetonico si verifica a causa dell'azione di massa: un abbondante apporto di AcAc e il rapido consumo di acetil-CoA attraverso la citrato sintasi favoriscono la formazione di AcAc-CoA (+ succinato) da parte di SCOT. In particolare, a differenza del glucosio (esochinasi) e degli acidi grassi (sintetasi acil-CoA), l'attivazione dei corpi chetonici (SCOT) in una forma ossidabile non richiede l'investimento di ATP. Una reazione reversibile della tiolasi AcAc-CoA [catalizzata da una qualsiasi delle quattro tiolasi mitocondriali codificate da ACAA2 (che codifica un enzima noto come T1 o CT), ACAT1 (codifica T2), HADHA o HADHB] produce due molecole di acetil-CoA, che entrano nel ciclo TCA (Hersh e Jencks, 1967; Stern et al., 1956; Williamson et al., 1971). Durante gli stati chetotici (cioè, chetoni sierici totali> 500 M), i corpi chetonici contribuiscono in modo significativo al dispendio energetico e vengono utilizzati rapidamente nei tessuti fino a quando si verifica l'assorbimento o la saturazione dell'ossidazione (Balasse et al., 1978; Balasse e Fery, 1989 ; Edmond et al., 1987). Una frazione molto piccola dei corpi chetonici derivati dal fegato può essere prontamente misurata nelle urine e le velocità di utilizzo e riassorbimento da parte del rene sono proporzionate alla concentrazione circolante (Goldstein, 1987; Robinson e Williamson, 1980). Durante gli stati altamente chetotici (> 1 mM nel plasma), la chetonuria funge da reporter semiquantitativo della chetosi, sebbene la maggior parte dei test clinici sui corpi chetonici delle urine rilevi AcAc ma non? OHB (Klocker et al., 2013).
Substrati chetogenici e loro impatto sul metabolismo degli epatociti
I substrati chetogenici comprendono acidi grassi e amminoacidi (Fig. 1B). Il catabolismo degli amminoacidi, in particolare la leucina, genera circa il 4% dei corpi chetonici nello stato postassorbente (Thomas et al., 1982). Pertanto il pool di substrati dell'acetile-CoA per generare corpi chetonici deriva principalmente da acidi grassi, poiché durante gli stati di ridotta assunzione di carboidrati, il piruvato entra nel ciclo epatico del TCA principalmente tramite anaplerosi, cioè carbossilazione ATP-dipendente a ossalacetato (OAA) o malato (MAL) e non decarbossilazione ossidativa in acetil-CoA (Jeoung et al., 2012; Magnusson et al., 1991; Merritt et al., 2011). Nel fegato, il glucosio e il piruvato contribuiscono in modo trascurabile alla chetogenesi, anche quando la piruvato decarbossilazione in acetil-CoA è massima (Jeoung et al., 2012).
L'acetil-CoA ricopre diversi ruoli integrali del metabolismo intermedio epatico oltre la generazione di ATP attraverso l'ossidazione terminale (vedi anche Integrazione del metabolismo del corpo chetone, modificazione post-traduzionale e fisiologia cellulare). L'acetil-CoA attiva allostericamente (i) piruvato carbossilasi (PC), attivando così un meccanismo di controllo metabolico che aumenta l'ingresso anaplerotico dei metaboliti nel ciclo TCA (Owen et al., 2002; Scrutton e Utter, 1967) e (ii) piruvato deidrogenasi chinasi, che fosforila e inibisce la piruvato deidrogenasi (PDH) (Cooper et al., 1975), migliorando così ulteriormente il flusso di piruvato nel ciclo TCA attraverso l'anaplerosi. Inoltre, l'acetil-CoA citoplasmatico, il cui pool è potenziato da meccanismi che convertono l'acetil-CoA mitocondriale in metaboliti trasportabili, inibisce l'ossidazione degli acidi grassi: acetil-CoA carbossilasi (ACC) catalizza la conversione di acetil-CoA in malonil-CoA, il substrato lipogenico e inibitore allosterico del CPT1 mitocondriale [rivisto in (Kahn et al., 2005; McGarry and Foster, 1980)]. Pertanto, il pool di acetil-CoA mitocondriale regola sia ed è regolato dalla via di spillover della chetogenesi, che orchestra aspetti chiave del metabolismo intermedio epatico.
Fatti metabolici non ossidativi dei corpi chetonici
Il destino predominante dei chetoni derivati dal fegato è l'ossidazione extraepatica dipendente dallo SCOT. Tuttavia, AcAc può essere esportato dai mitocondri e utilizzato in percorsi anabolici attraverso la conversione in AcAc-CoA mediante una reazione dipendente dall'ATP catalizzata dalla citetilmetano acetoacetil-CoA sintetasi (AACS, Fig. 1B). Questo percorso è attivo durante lo sviluppo del cervello e nella ghiandola mammaria che allatta (Morris, 2005, Robinson and Williamson, 1978; Ohgami et al., 2003). L'AACS è anche altamente espresso nel tessuto adiposo e negli osteoclasti attivati (Aguilo et al., 2010; Yamasaki et al., 2016). AcAc-CoA citoplasmatico può essere diretto da citosolico HMGCS1 verso la biosintesi dello sterolo, o scisso da due di due tiolasi citoplasmatiche ad acetil-CoA (ACAA1 e ACAT2), carbossilato a malonil-CoA e contribuire alla sintesi di acidi grassi (Bergstrom et al., 1984; Edmond, 1974; Endemann et al., 1982; Geelen et al., 1983; Webber e Edmond, 1977).
Mentre il significato fisiologico deve ancora essere stabilito, i chetoni possono fungere da substrati anabolizzanti anche nel fegato. In contesti sperimentali artificiali, AcAc può contribuire fino alla metà dei lipidi di nuova sintesi e fino al 75% del nuovo colesterolo sintetizzato (Endemann et al., 1982; Geelen et al., 1983; Freed et al., 1988). Poiché AcAc deriva dall'ossidazione incompleta dei grassi epatici, la capacità di AcAc di contribuire alla lipogenesi in vivo implicherebbe un ciclo futile epatico, dove i chetoni derivati dal grasso possono essere utilizzati per la produzione di lipidi, una nozione il cui significato fisiologico richiede una convalida sperimentale, ma potrebbe servire ruoli adattivi o disadattivi (Solinas et al., 2015). AcAc fornisce avidamente la colesterogenesi, con un basso AACS Km-AcAc (~ 50 M) che favorisce l'attivazione di AcAc anche a stomaco pieno (Bergstrom et al., 1984). Il ruolo dinamico del metabolismo dei chetoni citoplasmatici è stato suggerito nei neuroni embrionali primari di topo e negli adipociti derivati 3T3-L1, poiché il knockdown dell'AACS ha alterato la differenziazione di ciascun tipo di cellula (Hasegawa et al., 2012a; Hasegawa et al., 2012b). Il knockdown dell'AACS nei topi in vivo ha ridotto il colesterolo sierico (Hasegawa et al., 2012c). SREBP-2, un regolatore trascrizionale principale della biosintesi del colesterolo e recettore attivato dal proliferatore del perossisoma (PPAR) -? sono attivatori trascrizionali dell'AACS e regolano la sua trascrizione durante lo sviluppo dei neuriti e nel fegato (Aguilo et al., 2010; Hasegawa et al., 2012c). Nel loro insieme, il metabolismo del corpo chetonico citoplasmatico può essere importante in determinate condizioni o storie naturali di malattie, ma è inadeguato per smaltire i corpi chetonici derivati dal fegato, poiché si verifica una massiccia iperchetonemia nel contesto di una compromissione selettiva del destino ossidativo primario attraverso la perdita di mutazioni funzionali a SCOT (Berry et al., 2001; Cotter et al., 2011).
Regolazione di HMGCS2 e SCOT / OXCT1
La divergenza di un mitocondrio dal gene che codifica HMGCS citosolico si è manifestata precocemente nell'evoluzione dei vertebrati a causa della necessità di supportare la chetogenesi epatica in specie con rapporti tra il cervello e il peso corporeo più elevati (Boukaftane et al., 1994; Cunnane and Crawford, 2003). Le mutazioni HMGCS2 di perdita naturale di funzione nell'uomo causano attacchi di ipoglicemia ipoketotica (Pitt et al., 2015; Thompson et al., 1997). L'espressione robusta HMGCS2 è limitata agli epatociti e all'epitelio del colon, e la sua espressione e attività enzimatica sono coordinate attraverso diversi meccanismi (Mascaro et al., 1995; McGarry e Foster, 1980; Robinson e Williamson, 1980). Mentre l'intera gamma di stati fisiologici che influenzano HMGCS2 richiede un'ulteriore delucidazione, la sua espressione e / o attività è regolata durante il periodo postnatale, invecchiamento, diabete, fame o ingestione di dieta chetogenica (Balasse e Fery, 1989; Cahill GF Jr, 2006 ; Girard et al., 1992; Hegardt, 1999; Satapati et al., 2012; Sengupta et al., 2010). Nel feto, la metilazione della regione fiancheggiante 5 del gene Hmgcs2 è inversamente correlata alla sua trascrizione ed è parzialmente invertita dopo la nascita (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al. ., 1983). Allo stesso modo, il Bdh1 epatico mostra un pattern di espressione evolutivo, crescente dalla nascita allo svezzamento, ed è anche indotto dalla dieta chetogenica in un fattore di crescita dei fibroblasti (FGF) -21-dipendente (Badman et al., 2007; Zhang et al., 1989 ). La chetogenesi nei mammiferi è altamente reattiva sia per l'insulina che per il glucagone, essendo soppressa e stimolata, rispettivamente (McGarry e Foster, 1977). L'insulina sopprime la lipolisi del tessuto adiposo, privando così la chetogenesi del suo substrato, mentre il glucagone aumenta il flusso chetogenico attraverso un effetto diretto sul fegato (Hegardt, 1999). La trascrizione di Hmgcs2 è stimolata dal fattore di trascrizione forkhead FOXA2, che viene inibito tramite insulina-fosfatidilinositolo-3-chinasi / Akt ed è indotto dal segnale glucagone-cAMP-p300 (Arias et al., 1995; Hegardt, 1999; Quant et al. , 1990; Thumelin et al., 1993; von Meyenn et al., 2013; Wolfrum et al., 2004; Wolfrum et al., 2003). PPAR? (Rodriguez et al., 1994) insieme al suo target, FGF21 (Badman et al., 2007) inducono anche la trascrizione di Hmgcs2 nel fegato durante la fame o la somministrazione di dieta chetogenica (Badman et al., 2007; Inagaki et al., 2007 ). Induzione di PPAR? può verificarsi prima del passaggio dalla fisiologia fetale a quella neonatale, mentre l'attivazione di FGF21 può essere favorita nel periodo neonatale precoce tramite l'inibizione mediata da? OHB dell'istone deacetilasi (HDAC) -3 (Rando et al., 2016). Inibizione dipendente da mTORC1 (mammifero bersaglio del complesso rapamicina 1) di PPAR? l'attività trascrizionale è anche un regolatore chiave dell'espressione genica di Hmgcs2 (Sengupta et al., 2010) e il PER2 epatico, un oscillatore circadiano principale, regola indirettamente l'espressione di Hmgcs2 (Chavan et al., 2016). Recenti osservazioni indicano che l'interleuchina-6 extraepatica indotta dal tumore altera la chetogenesi tramite PPAR? soppressione (Flint et al., 2016).
L'attività dell'enzima HMGCS2 è regolata tramite più PTM. La fosforilazione di serina HMGCS2 ha potenziato la sua attività in vitro (Grimsrud et al., 2012). L'attività di HMGCS2 è inibita allostericamente dalla succinilazione di succinil-CoA e residui di lisina (Arias et al., 1995; Hegardt, 1999; Lowe and Tubbs, 1985; Quant et al., 1990; Rardin et al., 2013; Reed et al., 1975; Thumelin et al., 1993). La succinilazione dei residui di lisina HMGCS2, HMGCL e BDH1 nei mitocondri epatici sono bersagli del deacilasi sirtuin dipendente da NAD + 5 (SIRT5) (Rardin et al., 2013). L'attività di HMGCS2 è anche potenziata dalla deacetilazione della lisina SIRT3 ed è possibile che la diafonia tra acetilazione e succinilazione regoli l'attività di HMGCS2 (Rardin et al., 2013; Shimazu et al., 2013). Nonostante la capacità di questi PTM di regolare HMGCS2 Km e Vmax, le fluttuazioni di questi PTM non sono state ancora accuratamente mappate e non sono state confermate come driver meccanicistici della chetogenesi in vivo.
SCOT è espresso in tutte le cellule di mammiferi che ospitano i mitocondri, ad eccezione di quelli degli epatociti. L'importanza dell'attività di SCOT e della chetolisi è stata dimostrata nei topi SCOT-KO, che hanno presentato una letalità uniforme a causa dell'ipoglicemia iperchetonemia entro 48h dopo la nascita (Cotter et al., 2011). La perdita di SCOT dei tessuti nei neuroni o nei miociti scheletrici induce anomalie metaboliche durante la fame ma non è letale (Cotter et al., 2013b). Nell'uomo il deficit di SCOT si manifesta precocemente nella vita con chetoacidosi grave, che causa letargia, vomito e coma (Berry et al., 2001; Fukao et al., 2000; Kassovska-Bratinova et al., 1996; Niezen-Koning et al. , 1997; Saudubray et al., 1987; Snyderman et al., 1998; Tildon e Cornblath, 1972). Relativamente poco è noto a livello cellulare sui regolatori di espressione di geni e proteine SCOT. L'espressione dell'mRNA di Oxct1 e la proteina e l'attività di SCOT sono diminuite negli stati chetotici, possibilmente attraverso meccanismi PPAR-dipendenti (Fenselau e Wallis, 1974; Fenselau e Wallis, 1976; Grinblat et al., 1986; Okuda et al., 1991; Turko et al ., 2001; Wentz et al., 2010). Nella chetoacidosi diabetica, la mancata corrispondenza tra chetogenesi epatica ed ossidazione extraepatica si acuisce a causa della compromissione dell'attività di SCOT. La sovraespressione del trasportatore di glucosio insulino-indipendente (GLUT1 / SLC2A1) nei cardiomiociti inibisce anche l'espressione del gene Oxct1 e downregula l'ossidazione terminale dei chetoni in uno stato non chetotico (Yan et al., 2009). Nel fegato, l'abbondanza di mRNA di Oxct1 è soppressa da microRNA-122 e metilazione dell'istone H3K27me3 che sono evidenti durante la transizione dal periodo fetale a quello neonatale (Thorrez et al., 2011). Tuttavia, la soppressione dell'espressione di Oxct1 epatica nel periodo postnatale è principalmente attribuibile all'evacuazione di progenitori ematopoietici che esprimono Oxct1 dal fegato, piuttosto che alla perdita dell'espressione di Oxct1 preesistente in epatociti differenziati terminalmente. Infatti, l'espressione dell'mRNA di Oxct1 e della proteina SCOT negli epatociti differenziati è estremamente bassa (Orii et al., 2008).
SCOT è anche regolamentato da PTM. L'enzima è iperacetilato nel cervello dei topi SIRT3 KO, che mostrano anche una ridotta produzione di acetil-CoA dipendente da AcAc (Dittenhafer-Reed et al., 2015). La nitrazione non enzimatica dei residui di tirosina di SCOT ne attenua anche l'attività, che è stata segnalata nei cuori di vari modelli di topi diabetici (Marcondes et al., 2001; Turko et al., 2001; Wang et al., 2010a). Al contrario, la nitrazione dei residui di triptofano aumenta l'attività degli SCOT (Brégére et al., 2010; Rebrin et al., 2007). Possono esistere meccanismi molecolari di nitrazione o de-nitrazione specifici del residuo progettati per modulare l'attività SCOT e richiedono delucidazione.
Controversie nella chetogenesi extraepatica
Nei mammiferi l'organo chetogenico primario è il fegato e solo gli epatociti e le cellule epiteliali intestinali esprimono abbondantemente l'isoforma mitocondriale di HMGCS2 (Cotter et al., 2013a; Cotter et al., 2014; McGarry e Foster, 1980; Robinson e Williamson, 1980) . La fermentazione batterica anaerobica di polisaccaridi complessi produce butirrato, che viene assorbito dai colonociti nei mammiferi per ossidazione terminale o chetogenesi (Cherbuy et al., 1995), che può svolgere un ruolo nella differenziazione dei colonociti (Wang et al., 2016). Escludendo le cellule epiteliali intestinali e gli epatociti, l'HMGCS2 è quasi assente in quasi tutte le altre cellule di mammifero, ma la prospettiva di chetogenesi extraepatica è stata sollevata nelle cellule tumorali, negli astrociti del sistema nervoso centrale, nel rene, nel pancreas? cellule, epitelio pigmentato retinico (RPE) e persino nel muscolo scheletrico (Adijanto et al., 2014; Avogaro et al., 1992; El Azzouny et al., 2016; Grabacka et al., 2016; Kang et al., 2015 ; Le Foll et al., 2014; Nonaka et al., 2016; Takagi et al., 2016a; Thevenet et al., 2016; Zhang et al., 2011). HMGCS2 ectopico è stato osservato in tessuti privi di capacità chetogenica netta (Cook et al., 2016; Wentz et al., 2010) e HMGCS2 mostra attività prospettiche di `` moonlighting '' indipendenti dalla chetogenesi, anche all'interno del nucleo cellulare (Chen et al. , 2016; Kostiuk et al., 2010; Meertens et al., 1998).
Qualsiasi tessuto extraepatico che ossida i corpi chetonici ha anche il potenziale per accumulare corpi chetonici tramite meccanismi indipendenti HMGCS2 (Fig. 2A). Tuttavia, non esiste tessuto extraepatico in cui una concentrazione corporea di chetoni allo stato stazionario superi quella nella circolazione (Cotter et al., 2011; Cotter et al., 2013b; Harrison e Long, 1940), sottolineando che i corpi chetonici vengono trasportati gradiente di concentrazione tramite meccanismi MCT1 / 2-dipendenti. Un meccanismo di apparente chetogenesi extraepatica può effettivamente riflettere una relativa compromissione dell'ossidazione chetonica. Ulteriori potenziali spiegazioni rientrano nel regno della formazione del corpo chetonico. In primo luogo, la chetogenesi de novo può verificarsi tramite l'attività enzimatica reversibile della tiolasi e della SCOT (Weidemann e Krebs, 1969). Quando la concentrazione di acetil-CoA è relativamente alta, le reazioni normalmente responsabili dell'ossidazione di AcAc operano nella direzione opposta (GOLDMAN, 1954). Un secondo meccanismo si verifica quando gli intermedi derivati dall'ossidazione si accumulano a causa di un collo di bottiglia del ciclo TCA, AcAc-CoA viene convertito in l-? OHB-CoA attraverso una reazione catalizzata dalla 3-idrossiacil-CoA deidrogenasi mitocondriale e ulteriormente da 3-idrossibutirile CoA deacylase a l-? OHB, che è indistinguibile mediante spettrometria di massa o spettroscopia di risonanza dall'enantiomero fisiologico d-? OHB (Reed e Ozand, 1980). l-? OHB può essere distinto cromatograficamente o enzimaticamente da d-? OHB ed è presente nei tessuti extraepatici, ma non nel fegato o nel sangue (Hsu et al., 2011). La chetogenesi epatica produce solo d-? OHB, l'unico enantiomero che è un substrato BDH (Ito et al., 1984; Lincoln et al., 1987; Reed and Ozand, 1980; Scofield et al., 1982; Scofield et al., 1982). Un terzo meccanismo indipendente da HMGCS2 genera d-? OHB attraverso il catabolismo degli amminoacidi, in particolare quello della leucina e della lisina. Un quarto meccanismo è apparente solo perché è dovuto a un artefatto di etichettatura ed è quindi definito pseudoketogenesis. Questo fenomeno è attribuibile alla reversibilità delle reazioni SCOT e tiolasi e può causare una sovrastima del turnover corporeo chetonico a causa della diluizione isotopica del tracciante corporeo chetonico nel tessuto extraepatico (Des Rosiers et al., 1990; Fink et al., 1988) . Tuttavia, la pseudochetogenesi può essere trascurabile nella maggior parte dei contesti (Bailey et al., 1990; Keller et al., 1978). Uno schema (Fig. 2A) indica un approccio utile da applicare considerando un'elevata concentrazione tissutale di chetoni allo stato stazionario.


Il rene è stato recentemente oggetto di attenzione come organo potenzialmente chetogenico. Nella stragrande maggioranza degli stati, il rene è un consumatore netto di corpi chetonici derivati dal fegato, che espellono o riassorbono corpi chetonici dal flusso sanguigno, e il rene generalmente non è un generatore o concentratore di corpi chetonici netti (Robinson e Williamson, 1980). Gli autori di uno studio classico hanno concluso che la chetogenesi renale minima quantificata in un sistema sperimentale artificiale non era fisiologicamente rilevante (Weidemann e Krebs, 1969). Recentemente, la chetogenesi renale è stata dedotta in modelli murini diabetici e con deficit di autofagia, ma è più probabile che cambiamenti multiorgano nell'omeostasi metabolica alterino il metabolismo chetonico integrativo attraverso input su più organi (Takagi et al., 2016a; Takagi et al., 2016b; Zhang et al., 2011). Una recente pubblicazione ha suggerito la chetogenesi renale come meccanismo protettivo contro il danno da ischemia-riperfusione nel rene (Tran et al., 2016). Le concentrazioni assolute allo stato stazionario di? OHB da estratti di tessuto renale di topo sono state riportate a ~ 4-12 mM. Per verificare se questo era sostenibile, abbiamo quantificato le concentrazioni di? OHB in estratti renali da topi alimentati e 24 ore a digiuno. Le concentrazioni di? OHB nel siero sono aumentate da ~ 100 M a 2 mM con 24 ore di digiuno (Fig. 2B), mentre le concentrazioni renali di? OHB allo stato stazionario si avvicinano a 100 M a stomaco pieno e solo 1 mM a 24 ore di digiuno (Fig. 2C E), osservazioni coerenti con le concentrazioni quantificate oltre 45 anni fa (Hems e Brosnan, 1970). Resta possibile che negli stati chetotici, i corpi chetonici derivati dal fegato possano essere renoprotettivi, ma l'evidenza della chetogenesi renale richiede ulteriori prove. Prove convincenti che supportano la vera chetogenesi extraepatica sono state presentate in RPE (Adijanto et al., 2014). Questa intrigante trasformazione metabolica è stata suggerita per consentire potenzialmente ai chetoni derivati dall'RPE di fluire al fotorecettore o alle cellule della glia di Mller, che potrebbero aiutare nella rigenerazione del segmento esterno del fotorecettore.
? OHB come mediatore di segnalazione
Sebbene siano energeticamente ricchi, i corpi chetonici esercitano ruoli di segnalazione provocatori `` non canonici '' nell'omeostasi cellulare (Fig.3) (Newman e Verdin, 2014; Rojas-Morales et al., 2016). Ad esempio,? OHB inibisce gli HDAC di Classe I, che aumenta l'acetilazione degli istoni e quindi induce l'espressione di geni che riducono lo stress ossidativo (Shimazu et al., 2013). ? L'OHB stesso è un modificatore covalente dell'istone dei residui di lisina nel fegato di topi diabetici a digiuno o indotti da streptozotocina (Xie et al., 2016) (vedere anche di seguito, Integrazione del metabolismo corporeo chetonico, modificazione post-traduzionale e fisiologia cellulare, e Corpi chetonici, stress ossidativo e neuroprotezione).

? L'OHB è anche un effettore tramite i recettori accoppiati alla proteina G. Attraverso meccanismi molecolari poco chiari, sopprime l'attività del sistema nervoso simpatico e riduce il dispendio energetico totale e la frequenza cardiaca inibendo la segnalazione degli acidi grassi a catena corta attraverso il recettore accoppiato alla proteina G 41 (GPR41) (Kimura et al., 2011). Uno degli effetti di segnalazione più studiati di? OHB procede attraverso GPR109A (noto anche come HCAR2), un membro della sottofamiglia GPCR dell'acido idrocarbossilico espresso nei tessuti adiposi (bianchi e marroni) (Tunaru et al., 2003), e in cellule immunitarie (Ahmed et al., 2009). ? OHB è l'unico ligando endogeno noto del recettore GPR109A (EC50 ~ 770 M) attivato da d-? OHB, l-? OHB e butirrato, ma non AcAc (Taggart et al., 2005). L'elevata soglia di concentrazione per l'attivazione di GPR109A viene raggiunta attraverso l'adesione a una dieta chetogenica, fame o durante la chetoacidosi, portando all'inibizione della lipolisi del tessuto adiposo. L'effetto anti-lipolitico di GPR109A procede attraverso l'inibizione dell'adenil ciclasi e della diminuzione del cAMP, inibendo la lipasi trigliceridica sensibile agli ormoni (Ahmed et al., 2009; Tunaru et al., 2003). Questo crea un ciclo di feedback negativo in cui la chetosi pone un freno modulatorio alla chetogenesi diminuendo il rilascio di acidi grassi non esterificati dagli adipociti (Ahmed et al., 2009; Taggart et al., 2005), un effetto che può essere controbilanciato da la spinta simpatica che stimola la lipolisi. La niacina (vitamina B3, acido nicotinico) è un potente ligando (EC50 ~ 0.1 M) per GRP109A, efficacemente impiegato per decenni per le dislipidemie (Benyo et al., 2005; Benyo et al., 2006; Fabbrini et al., 2010a; Lukasova et al., 2011; Tunaru et al., 2003). Mentre la niacina aumenta il trasporto inverso del colesterolo nei macrofagi e riduce le lesioni aterosclerotiche (Lukasova et al., 2011), gli effetti di? OHB sulle lesioni aterosclerotiche rimangono sconosciuti. Sebbene il recettore GPR109A eserciti ruoli protettivi ed esistano connessioni intriganti tra l'uso della dieta chetogenica nell'ictus e nelle malattie neurodegenerative (Fu et al., 2015; Rahman et al., 2014), un ruolo protettivo di? OHB tramite GPR109A non è stato dimostrato in vivo .
Infine,? OHB può influenzare l'appetito e la sazietà. Una meta-analisi di studi che hanno misurato gli effetti delle diete chetogeniche ea bassissimo contenuto energetico ha concluso che i partecipanti che consumano queste diete mostrano una maggiore sazietà, rispetto alle diete di controllo (Gibson et al., 2015). Tuttavia, una spiegazione plausibile per questo effetto sono gli elementi metabolici o ormonali aggiuntivi che potrebbero modulare l'appetito. Ad esempio, i topi mantenuti con una dieta chetogenica di roditori hanno mostrato un aumento del dispendio energetico rispetto ai topi nutriti con chow control, nonostante un apporto calorico simile, e la leptina circolante oi geni dei peptidi che regolano il comportamento alimentare non sono stati modificati (Kennedy et al., 2007). Tra i meccanismi proposti che suggeriscono la soppressione dell'appetito da? OHB include sia la segnalazione che l'ossidazione (Laeger et al., 2010). La delezione specifica degli epatociti del gene del ritmo circadiano (Per2) e studi di immunoprecipitazione della cromatina hanno rivelato che PER2 attiva direttamente il gene Cpt1a e regola indirettamente Hmgcs2, portando a chetosi alterata nei topi knockout per Per2 (Chavan et al., 2016). Questi topi hanno mostrato una ridotta anticipazione del cibo, che è stata parzialmente ripristinata dalla somministrazione sistemica di? OHB. Saranno necessari studi futuri per confermare il sistema nervoso centrale come bersaglio diretto? OHB e se l'ossidazione chetonica è necessaria per gli effetti osservati, o se è coinvolto un altro meccanismo di segnalazione. Altri ricercatori hanno invocato la possibilità di una chetogenesi derivata dagli astrociti locale all'interno dell'ipotalamo ventromediale come regolatore dell'assunzione di cibo, ma queste osservazioni preliminari beneficeranno anche di valutazioni genetiche e basate sul flusso (Le Foll et al., 2014). La relazione tra chetosi e mancanza di nutrienti rimane interessante perché la fame e la sazietà sono elementi importanti nei tentativi di perdita di peso falliti.
Integrazione del metabolismo corporeo chetonico, modificazione post-traduzionale e fisiologia cellulare
I corpi chetonici contribuiscono a compartimenti stagni di acetil-CoA, un intermedio chiave che presenta ruoli importanti nel metabolismo cellulare (Pietrocola et al., 2015). Un ruolo dell'acetil-CoA è quello di fungere da substrato per l'acetilazione, una modifica covalente istonica enzimaticamente catalizzata (Choudhary et al., 2014; Dutta et al., 2016; Fan et al., 2015; Menzies et al., 2016 ). Un gran numero di proteine mitocondriali dinamicamente acetilate, molte delle quali possono verificarsi attraverso meccanismi non enzimatici, sono emerse anche da studi di proteomica computazionale (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013 Shimazu et al., 2010). Le deacetilasi di lisina usano un cofattore di zinco (ad esempio, HDAC nucleocitosolici) o NAD + come co-substrato (sirtuine, SIRT) (Choudhary et al., 2014; Menzies et al., 2016). L'acetilproteoma funge sia da sensore che da effettore del pool di acetil-CoA cellulare totale, poiché le manipolazioni fisiologiche e genetiche portano ciascuna a variazioni globali non enzimatiche di acetilazione (Weinert et al., 2014). Poiché i metaboliti intracellulari fungono da modulatori dell'acetilazione dei residui di lisina, è importante considerare il ruolo dei corpi chetonici, la cui abbondanza è altamente dinamica.
? OHB è un modificatore epigenetico attraverso almeno due meccanismi. Livelli aumentati di? OHB indotti da digiuno, restrizione calorica, somministrazione diretta o esercizio prolungato provocano l'inibizione di HDAC o l'attivazione dell'istone acetiltransferasi (Marosi et al., 2016; Sleiman et al., 2016) o allo stress ossidativo (Shimazu et al., 2013) . ? L'inibizione dell'OHB di HDAC3 potrebbe regolare la fisiologia metabolica del neonato (Rando et al., 2016). Indipendentemente, lo stesso? OHB modifica direttamente i residui di lisina istonica (Xie et al., 2016). Il digiuno prolungato o la chetoacidosi diabetica indotta dalla steptozotocina hanno aumentato l'istone? -Idrossibutirilazione. Sebbene il numero di siti di lisina-idrossibutirilazione e acetilazione fosse paragonabile, è stata osservata stechiometricamente maggiore istone-idrossibutirilazione rispetto all'acetilazione. Geni distinti sono stati influenzati dall'istone lisina? -Idrossibutirilazione, rispetto all'acetilazione o alla metilazione, suggerendo funzioni cellulari distinte. Non è noto se la? -Idrossibutirilazione sia spontanea o enzimatica, ma espande la gamma di meccanismi attraverso i corpi chetonici che influenzano dinamicamente la trascrizione.
Gli eventi di riprogrammazione cellulare essenziale durante la restrizione calorica e la privazione dei nutrienti possono essere mediati nella deacetilazione mitocondriale e nella desuccinilazione dipendenti da SIRT3 e SIRT5, rispettivamente, regolando le proteine chetogeniche e chetolitiche a livello post-traduzionale nel fegato e nei tessuti extraepatici (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013; Shimazu et al., 2010). Anche se il confronto stechiometrico dei siti occupati non si collega necessariamente direttamente ai cambiamenti nel flusso metabolico, l'acetilazione mitocondriale è dinamica e può essere guidata dalla concentrazione di acetil-CoA o dal pH mitocondriale, piuttosto che dalle acetiltransferasi enzimatiche (Wagner e Payne, 2013). Il fatto che SIRT3 e SIRT5 modulino le attività degli enzimi che metabolizzano il corpo chetonico provoca la questione del ruolo reciproco dei chetoni nello scolpire l'acetilproteoma, il succinilproteoma e altri bersagli cellulari dinamici. Infatti, poiché le variazioni della chetogenesi riflettono le concentrazioni di NAD +, la produzione e l'abbondanza di chetoni potrebbero regolare l'attività della sirtuina, influenzando così i pool totali di acetil-CoA / succinil-CoA, l'acilproteoma e quindi la fisiologia mitocondriale e cellulare. ? -idrossibutirilazione dei residui di lisina enzimatica potrebbe aggiungere un altro strato alla riprogrammazione cellulare. Nei tessuti extraepatici, l'ossidazione del corpo chetonico può stimolare analoghi cambiamenti nell'omeostasi cellulare. Mentre la compartimentazione dei pool di acetil-CoA è altamente regolata e coordina un ampio spettro di cambiamenti cellulari, la capacità dei corpi chetonici di modellare direttamente le concentrazioni di acetil-CoA mitocondriali e citoplasmatiche richiede delucidazione (Chen et al., 2012; Corbet et al., 2016; Pougovkina et al., 2014; Schwer et al., 2009; Wellen e Thompson, 2012). Poiché le concentrazioni di acetil-CoA sono strettamente regolate e l'acetil-CoA è impermeabile alla membrana, è fondamentale considerare i meccanismi driver che coordinano l'omeostasi dell'acetil-CoA, inclusi i tassi di produzione e l'ossidazione terminale nel ciclo TCA, la conversione in corpi chetonici, mitocondriale efflusso tramite carnitina acetiltransferasi (CrAT) o esportazione di acetil-CoA in citosol dopo la conversione in citrato e il rilascio da parte dell'ATP citrato liasi (ACLY). I ruoli chiave di questi ultimi meccanismi nell'acetilproteoma cellulare e nell'omeostasi richiedono una comprensione corrispondente dei ruoli della chetogenesi e dell'ossidazione chetonica (Das et al., 2015; McDonnell et al., 2016; Moussaieff et al., 2015; Overmyer et al., 2015; Seiler et al., 2014; Seiler et al., 2015; Wellen et al., 2009; Wellen e Thompson, 2012). Saranno necessarie tecnologie convergenti in metabolomica e acilproteomica nell'impostazione di modelli geneticamente manipolati per specificare obiettivi e risultati.
Risposte anti- e pro-infiammatorie ai corpi chetonici
La chetosi e i corpi chetonici modulano l'infiammazione e la funzione delle cellule immunitarie, ma sono stati proposti meccanismi vari e persino discrepanti. La privazione prolungata di nutrienti riduce l'infiammazione (Youm et al., 2015), ma la chetosi cronica del diabete di tipo 1 è uno stato pro-infiammatorio (Jain et al., 2002; Kanikarla-Marie e Jain, 2015; Kurepa et al., 2012 ). I ruoli di segnalazione basati sul meccanismo per? OHB nell'infiammazione emergono perché molte cellule del sistema immunitario, inclusi macrofagi o monociti, esprimono abbondantemente GPR109A. Mentre? OHB esercita una risposta prevalentemente antinfiammatoria (Fu et al., 2014; Gambhir et al., 2012; Rahman et al., 2014; Youm et al., 2015), alte concentrazioni di corpi chetonici, in particolare AcAc, possono innescare una risposta pro-infiammatoria (Jain et al., 2002; Kanikarla-Marie e Jain, 2015; Kurepa et al., 2012).
I ruoli antinfiammatori dei ligandi GPR109A nell'aterosclerosi, nell'obesità, nelle malattie infiammatorie intestinali, nelle malattie neurologiche e nel cancro sono stati esaminati (Graff et al., 2016). L'espressione di GPR109A è aumentata nelle cellule RPE di modelli diabetici, pazienti diabetici umani (Gambhir et al., 2012) e nella microglia durante la neurodegenerazione (Fu et al., 2014). Gli effetti anti-infiammatori di? OHB sono potenziati dalla sovraespressione di GPR109A nelle cellule RPE e abrogati dall'inibizione farmacologica o dal knockout genetico di GPR109A (Gambhir et al., 2012). ? OHB e acido nicotinico esogeno (Taggart et al., 2005), entrambi conferiscono effetti antinfiammatori nel TNF? o infiammazione indotta da LPS diminuendo i livelli di proteine pro-infiammatorie (iNOS, COX-2) o citochine secrete (TNF ?, IL-1 ?, IL-6, CCL2 / MCP-1), in parte attraverso l'inibizione di NF -? B traslocazione (Fu et al., 2014; Gambhir et al., 2012). ? L'OHB riduce lo stress ER e l'inflammasoma NLRP3, attivando la risposta allo stress antiossidante (Bae et al., 2016; Youm et al., 2015). Tuttavia, nell'infiammazione neurodegenerativa, la protezione mediata da? OHB dipendente da GPR109A non coinvolge mediatori infiammatori come la segnalazione della via MAPK (p. Es., ERK, JNK, p38) (Fu et al., 2014), ma può richiedere PGD1 COX-2-dipendente produzione (Rahman et al., 2014). È interessante che il macrofago GPR109A sia necessario per esercitare un effetto neuroprotettivo in un modello di ictus ischemico (Rahman et al., 2014), ma la capacità di? OHB di inibire l'inflammasoma NLRP3 nei macrofagi derivati dal midollo osseo è GPR109A indipendente (Youm et al. ., 2015). Sebbene la maggior parte degli studi colleghi? OHB agli effetti antinfiammatori,? OHB può essere pro-infiammatorio e aumentare i marcatori di perossidazione lipidica negli epatociti di vitello (Shi et al., 2014). Gli effetti anti-infiammatori di? OHB possono quindi dipendere dal tipo di cellula, dalla concentrazione di? OHB, dalla durata dell'esposizione e dalla presenza o assenza di co-modulatori.
A differenza di? OHB, AcAc può attivare la segnalazione pro-infiammatoria. AcAc elevata, specialmente con un'alta concentrazione di glucosio, intensifica il danno delle cellule endoteliali attraverso un meccanismo dipendente dalla NADPH ossidasi / stress ossidativo (Kanikarla-Marie e Jain, 2015). Elevate concentrazioni di AcAc nel cordone ombelicale di madri diabetiche erano correlate con un più alto tasso di ossidazione delle proteine e concentrazione di MCP-1 (Kurepa et al., 2012). L'alto AcAc nei pazienti diabetici era correlato al TNF? espressione (Jain et al., 2002) e AcAc, ma non? OHB, TNF indotta, espressione di MCP-1, accumulo di ROS e diminuzione del livello di cAMP nelle cellule monocite umane U937 (Jain et al., 2002; Kurepa et al ., 2012).
I fenomeni di segnalazione dipendenti dal corpo chetonico sono spesso innescati solo con concentrazioni corporee di chetoni elevate (> 5 mM) e, nel caso di molti studi che collegano i chetoni a effetti pro o antinfiammatori, attraverso meccanismi poco chiari. Inoltre, a causa degli effetti contraddittori di? OHB contro AcAc sull'infiammazione e della capacità del rapporto AcAc /? OHB di influenzare il potenziale redox mitocondriale, i migliori esperimenti che valutano i ruoli dei corpi chetonici sui fenotipi cellulari confrontano gli effetti di AcAc e? OHB in rapporti variabili e concentrazioni cumulative variabili [ad esempio, (Saito et al., 2016)]. Infine, AcAc può essere acquistato in commercio solo come sale di litio o come estere etilico che richiede l'idrolisi di base prima dell'uso. Il catione di litio induce indipendentemente cascate di trasduzione del segnale (Manji et al., 1995) e l'anione AcAc è labile. Infine, gli studi che utilizzano d / l-? OHB racemico possono essere confusi, poiché solo lo stereoisomero d-? OHB può essere ossidato ad AcAc, ma d-? OHB e l-? OHB possono entrambi segnalare attraverso GPR109A, inibire l'inflammasoma NLRP3, e servono come substrati lipogenici.
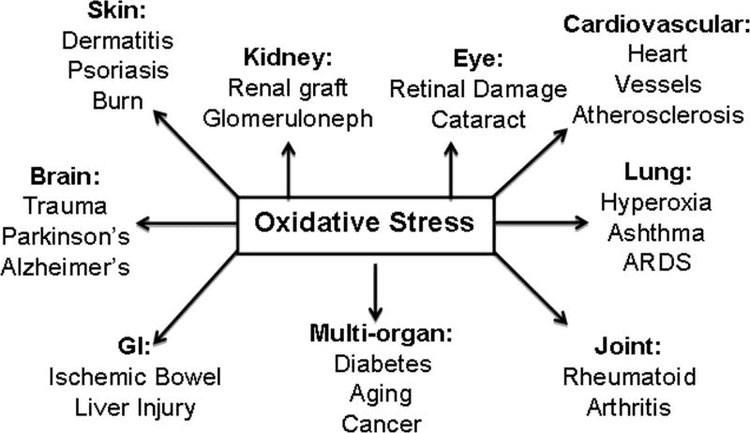
Corpi chetonici, stress ossidativo e neuroprotezione
Lo stress ossidativo è tipicamente definito come uno stato in cui i ROS si presentano in eccesso, a causa di un'eccessiva produzione e / o di una ridotta eliminazione. I ruoli antiossidanti e di mitigazione dello stress ossidativo dei corpi chetonici sono stati ampiamente descritti sia in vitro che in vivo, in particolare nel contesto della neuroprotezione. Poiché la maggior parte dei neuroni non genera efficacemente fosfati ad alta energia dagli acidi grassi, ma ossidano i corpi chetonici quando i carboidrati scarseggiano, gli effetti neuroprotettivi dei corpi chetonici sono particolarmente importanti (Cahill GF Jr, 2006; Edmond et al., 1987; Yang et al., 1987). Nei modelli di stress ossidativo, l'induzione BDH1 e la soppressione SCOT suggeriscono che il metabolismo del corpo chetone può essere riprogrammato per sostenere diversi segnali cellulari, potenziale redox o requisiti metabolici (Nagao et al., 2016; Tieu et al., 2003).
I corpi chetonici riducono i gradi di danno cellulare, lesioni, morte e apoptosi inferiore nei neuroni e nei cardiomiociti (Haces et al., 2008; Maalouf et al., 2007; Nagao et al., 2016; Tieu et al., 2003). I meccanismi invocati sono vari e non sempre correlati linearmente alla concentrazione. Basse concentrazioni millimolari di (do l) -? OHB eliminano ROS (anione idrossile), mentre AcAc elimina numerose specie di ROS, ma solo a concentrazioni che superano l'intervallo fisiologico (IC50 20 mM) (Haces et al., 67) . Al contrario, un'influenza benefica sul potenziale redox della catena di trasporto degli elettroni è un meccanismo comunemente collegato al d-? OHB. Mentre tutti e tre i corpi chetonici (d / l-? OHB e AcAc) hanno ridotto la morte delle cellule neuronali e l'accumulo di ROS innescato dall'inibizione chimica della glicolisi, solo d-? OHB e AcAc hanno prevenuto il declino dell'ATP neuronale. Al contrario, in un modello ipoglicemico in vivo, (do l) -? OHB, ma non AcAc ha impedito la perossidazione lipidica ippocampale (Haces et al., 2008; Maalouf et al., 2008; Marosi et al., 2007; Murphy, 2016 ; Tieu et al., 2009). Studi in vivo su topi alimentati con una dieta chetogenica (2003% kcal di grassi e 87% di proteine) hanno mostrato variazioni neuroanatomiche della capacità antiossidante (Ziegler et al., 13), dove i cambiamenti più profondi sono stati osservati nell'ippocampo, con aumento della glutatione perossidasi e totale capacità antiossidanti.
Dieta chetogenica, esteri chetonici (vedi anche Uso terapeutico di dieta chetogenica e corpi chetonici esogeni), o somministrazione di? OHB esercitano neuroprotezione in modelli di ictus ischemico (Rahman et al., 2014); Morbo di Parkinson (Tieu et al., 2003); sequestro di tossicità da ossigeno del sistema nervoso centrale (D'Agostino et al., 2013); spasmi epilettici (Yum et al., 2015); Encefalomiopatia mitocondriale, acidosi lattica e sindrome da episodi di ictus (MELAS) (Frey et al., 2016) e malattia di Alzheimer (Cunnane e Crawford, 2003; Yin et al., 2016). Al contrario, un recente rapporto ha dimostrato prove istopatologiche di progressione neurodegenerativa da una dieta chetogenica in un modello murino transgenico di riparazione anormale del DNA mitocondriale, nonostante l'aumento della biogenesi mitocondriale e delle firme antiossidanti (Lauritzen et al., 2016). Altri rapporti contrastanti suggeriscono che l'esposizione ad alte concentrazioni corporee di chetoni provoca stress ossidativo. Dosi elevate di? OHB o AcAc hanno indotto secrezione di ossido nitrico, perossidazione lipidica, ridotta espressione di SOD, glutatione perossidasi e catalasi negli epatociti di vitello, mentre negli epatociti di ratto l'induzione della via MAPK è stata attribuita ad AcAc ma non? OHB (Abdelmegeed et al., 2004 ; Shi et al., 2014; Shi et al., 2016).
Presi insieme, la maggior parte dei rapporti collega? OHB all'attenuazione dello stress ossidativo, poiché la sua somministrazione inibisce la produzione di ROS / superossido, previene la perossidazione lipidica e l'ossidazione delle proteine, aumenta i livelli di proteine antiossidanti e migliora la respirazione mitocondriale e la produzione di ATP (Abdelmegeed et al., 2004; Haces et al., 2008; Jain et al., 1998; Jain et al., 2002; Kanikarla-Marie e Jain, 2015; Maalouf et al., 2007; Maalouf e Rho, 2008; Marosi et al., 2016; Tieu et al., 2003; Yin et al., 2016; Ziegler et al., 2003). Sebbene l'AcAc sia stato correlato più direttamente di? OHB con l'induzione dello stress ossidativo, questi effetti non sono sempre facilmente sezionabili dalle risposte pro-infiammatorie prospettiche (Jain et al., 2002; Kanikarla-Marie e Jain, 2015; Kanikarla-Marie e Jain, 2016). Inoltre, è fondamentale considerare che l'apparente beneficio antiossidante conferito dalle diete chetogeniche pleiotropiche potrebbe non essere trasdotto dagli stessi corpi chetonici e la neuroprotezione conferita dai corpi chetonici potrebbe non essere interamente attribuibile allo stress ossidativo. Ad esempio, durante la privazione del glucosio, in un modello di deprivazione del glucosio nei neuroni corticali,? OHB ha stimolato il flusso autofagico e ha prevenuto l'accumulo di autofagosomi, che era associato a una diminuzione della morte neuronale (Camberos-Luna et al., 2016). d-? OHB induce anche le proteine antiossidanti canoniche FOXO3a, SOD, MnSOD e catalasi, prospetticamente attraverso l'inibizione di HDAC (Nagao et al., 2016; Shimazu et al., 2013).
Malattia del fegato grasso non alcolico (NAFLD) e metabolismo del corpo chetone
La NAFLD associata all'obesità e la steatoepatite non alcolica (NASH) sono le cause più comuni di malattia del fegato nei paesi occidentali (Rinella e Sanyal, 2016) e l'insufficienza epatica indotta dalla NASH è uno dei motivi più comuni per il trapianto di fegato. Mentre l'eccessivo stoccaggio di triacilgliceroli negli epatociti> 5% del peso del fegato (NAFL) da solo non causa una funzionalità epatica degenerativa, la progressione a NAFLD nell'uomo è correlata alla resistenza sistemica all'insulina e all'aumento del rischio di diabete di tipo 2 e può contribuire alla patogenesi del malattie cardiovascolari e malattie renali croniche (Fabbrini et al., 2009; Targher et al., 2010; Targher e Byrne, 2013). I meccanismi patogeni di NAFLD e NASH non sono completamente compresi ma includono anomalie del metabolismo degli epatociti, autofagia degli epatociti e stress del reticolo endoplasmatico, funzione delle cellule immunitarie epatiche, infiammazione del tessuto adiposo e mediatori infiammatori sistemici (Fabbrini et al., 2009; Masuoka e Chalasani, 2013 ; Targher et al., 2010; Yang et al., 2010). Le perturbazioni del metabolismo di carboidrati, lipidi e aminoacidi si verificano e contribuiscono all'obesità, al diabete e alla NAFLD negli esseri umani e negli organismi modello [rivisto in (Farese et al., 2012; Lin e Accili, 2011; Newgard, 2012; Samuel e Shulman, 2012; Sun e Lazar, 2013)]. Mentre le anomalie degli epatociti nel metabolismo dei lipidi citoplasmatici sono comunemente osservate nella NAFLD (Fabbrini et al., 2010b), il ruolo del metabolismo mitocondriale, che governa lo smaltimento ossidativo dei grassi è meno chiaro nella patogenesi della NAFLD. Anomalie del metabolismo mitocondriale si verificano e contribuiscono alla patogenesi NAFLD / NASH (Hyotylainen et al., 2016; Serviddio et al., 2011; Serviddio et al., 2008; Wei et al., 2008). C'è generale (Felig et al., 1974; Iozzo et al., 2010; Koliaki et al., 2015; Satapati et al., 2015; Satapati et al., 2012; Sunny et al., 2011) ma non uniforme ( Koliaki e Roden, 2013; Perry et al., 2016; Rector et al., 2010) consenso sul fatto che, prima dello sviluppo della NASH autentica, l'ossidazione mitocondriale epatica, e in particolare l'ossidazione dei grassi, è aumentata nell'obesità, insulino-resistenza sistemica e NAFLD. È probabile che con il progredire della NAFLD, emerga l'eterogenicità della capacità ossidativa, anche tra i singoli mitocondri, e alla fine la funzione ossidativa venga compromessa (Koliaki et al., 2015; Rector et al., 2010; Satapati et al., 2008; Satapati et al. ., 2012).
La chetogenesi viene spesso utilizzata come proxy per l'ossidazione dei grassi epatici. Le alterazioni della chetogenesi emergono man mano che la NAFLD progredisce nei modelli animali e probabilmente negli esseri umani. Attraverso meccanismi non completamente definiti, l'iperinsulinemia sopprime la chetogenesi, probabilmente contribuendo all'ipoketonemia rispetto ai controlli magri (Bergman et al., 2007; Bickerton et al., 2008; Satapati et al., 2012; Soeters et al., 2009; Sunny et al. , 2011; Vice et al., 2005). Tuttavia, la capacità della circolazione delle concentrazioni corporee di chetoni di predire la NAFLD è controversa (Mnnist et al., 2015; Sanyal et al., 2001). Robusti metodi spettroscopici di risonanza magnetica quantitativa in modelli animali hanno rivelato un aumento del tasso di turnover chetonico con moderata insulino-resistenza, ma tassi ridotti erano evidenti con una più grave resistenza all'insulina (Satapati et al., 2012; Sunny et al., 2010). Negli esseri umani obesi con fegato grasso, il tasso chetogenico è normale (Bickerton et al., 2008; Sunny et al., 2011), e quindi, i tassi di chetogenesi sono diminuiti rispetto all'aumento del carico di acidi grassi all'interno degli epatociti. Di conseguenza, l'acetil-CoA derivato dall'ossidazione può essere diretto all'ossidazione terminale nel ciclo TCA, aumentando l'ossidazione terminale, la gluconeogenesi guidata da fosfoenolpiruvato tramite anaplerosi / cataplerosi e stress ossidativo. L'acetil-CoA può anche essere esportato dai mitocondri come citrato, un substrato precursore per la lipogenesi (Fig.4) (Satapati et al., 2015; Satapati et al., 2012; Solinas et al., 2015). Mentre la chetogenesi diventa meno reattiva all'insulina o al digiuno con obesità prolungata (Satapati et al., 2012), i meccanismi sottostanti e le conseguenze a valle di ciò rimangono non completamente compresi. Prove recenti indicano che mTORC1 sopprime la chetogenesi in un modo che potrebbe essere a valle della segnalazione dell'insulina (Kucejova et al., 2016), che è concorde con le osservazioni secondo cui mTORC1 inibisce l'induzione di Hmgcs2 mediata da PPAR? (Sengupta et al., 2010) ( vedere anche il regolamento di HMGCS2 e SCOT / OXCT1).

Le osservazioni preliminari del nostro gruppo suggeriscono conseguenze epatiche avverse dell'insufficienza chetogenica (Cotter et al., 2014). Per verificare l'ipotesi che la chetogenesi alterata, anche in stati ricchi di carboidrati e quindi `` non chetogenici '', contribuisca al metabolismo del glucosio anormale e provochi la steatoepatite, abbiamo generato un modello murino di marcata insufficienza chetogenica mediante somministrazione di oligonucleotidi antisenso (ASO) mirati a Hmgcs2. La perdita di HMGCS2 nei topi adulti nutriti con cibo a basso contenuto di grassi standard ha causato una lieve iperglicemia e una produzione notevolmente aumentata di centinaia di metaboliti epatici, una suite dei quali ha fortemente suggerito l'attivazione della lipogenesi. L'alimentazione con dieta ad alto contenuto di grassi di topi con chetogenesi insufficiente ha provocato un esteso danno epatocitario e infiammazione. Questi risultati supportano l'ipotesi centrale che (i) la chetogenesi non sia una via di trabocco passivo ma piuttosto un nodo dinamico nell'omeostasi fisiologica epatica e integrata, e (ii) l'aumento chetogenico prudente per mitigare NAFLD / NASH e il metabolismo epatico disordinato del glucosio merita di essere esplorato .
In che modo la chetogenesi alterata può contribuire al danno epatico e all'alterazione dell'omeostasi del glucosio? La prima considerazione è se il colpevole sia la carenza di flusso chetogenico o gli stessi chetoni. Un recente rapporto suggerisce che i corpi chetonici possono mitigare il danno epatico indotto dallo stress ossidativo in risposta agli acidi grassi polinsaturi n-3 (Pawlak et al., 2015). Ricorda che a causa della mancanza di espressione di SCOT negli epatociti, i corpi chetonici non sono ossidati, ma possono contribuire alla lipogenesi e svolgere una varietà di ruoli di segnalazione indipendenti dalla loro ossidazione (vedi anche Destini metabolici non ossidativi dei corpi chetonici e? OHB come un mediatore di segnalazione). È anche possibile che i corpi chetonici derivati dagli epatociti possano servire come segnale e / o metabolita per i tipi di cellule adiacenti all'interno dell'acino epatico, comprese le cellule stellate e i macrofagi delle cellule di Kupffer. Mentre la letteratura limitata disponibile suggerisce che i macrofagi non sono in grado di ossidare i corpi chetonici, questo è stato misurato solo utilizzando metodologie classiche e solo nei macrofagi peritoneali (Newsholme et al., 1986; Newsholme et al., 1987), indicando che un ri- la valutazione è appropriata data l'abbondante espressione di SCOT nei macrofagi derivati dal midollo osseo (Youm et al., 2015).
Anche il flusso chetogenico degli epatociti può essere citoprotettivo. Sebbene i meccanismi salutari possano non dipendere dalla chetogenesi di per sé, le diete chetogeniche a basso contenuto di carboidrati sono state associate al miglioramento della NAFLD (Browning et al., 2011; Foster et al., 2010; Kani et al., 2014; Schugar e Crawford, 2012) . Le nostre osservazioni indicano che la chetogenesi degli epatociti può feedback e regolare il flusso del ciclo TCA, il flusso anaplerotico, la gluconeogenesi derivata dal fosfoenolpiruvato (Cotter et al., 2014) e persino il turnover del glicogeno. La compromissione chetogenica indirizza l'acetil-CoA ad aumentare il flusso di TCA, che nel fegato è stato collegato a un aumento del danno mediato da ROS (Satapati et al., 2015; Satapati et al., 2012); forza la diversione del carbonio in specie lipidiche sintetizzate de novo che potrebbero rivelarsi citotossiche; e previene la riossidazione del NADH a NAD + (Cotter et al., 2014) (Fig. 4). Presi insieme, sono necessari esperimenti futuri per affrontare i meccanismi attraverso i quali l'insufficienza chetogenica relativa può diventare disadattativa, contribuire all'iperglicemia, provocare steatoepatite e se questi meccanismi sono operativi nella NAFLD / NASH umana. Poiché l'evidenza epidemiologica suggerisce una chetogenesi compromessa durante la progressione della steatoepatite (Embade et al., 2016; Marinou et al., 2011; Mnnisté et al., 2015; Pramfalk et al., 2015; Safaei et al., 2016) le terapie che aumentano la chetogenesi epatica potrebbero rivelarsi salutari (Degirolamo et al., 2016; Honda et al., 2016).
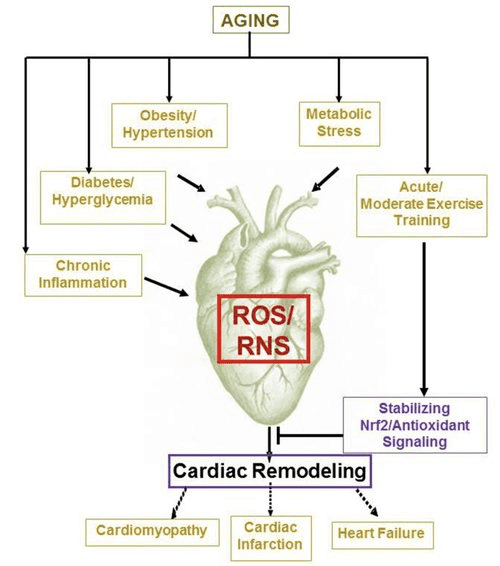
Corpi chetonici e insufficienza cardiaca (HF)
Con un tasso metabolico superiore a 400 kcal / kg / giorno e un turnover di 6 35 kg ATP / giorno, il cuore è l'organo con il più alto dispendio energetico e richiesta ossidativa (Ashrafian et al., 2007; Wang et al., 2010b). La stragrande maggioranza del ricambio energetico del miocardio risiede nei mitocondri e il 70% di questa fornitura proviene dalla FAO. Il cuore è onnivoro e flessibile in condizioni normali, ma il cuore che si rimodella patologicamente (p. Es., A causa di ipertensione o infarto miocardico) e il cuore diabetico diventano metabolicamente inflessibili (Balasse e Fery, 1989; BING, 1954; Fukao et al., 2004 ; Lopaschuk et al., 2010; Taegtmeyer et al., 1980; Taegtmeyer et al., 2002; Young et al., 2002). Infatti, le anomalie geneticamente programmate del metabolismo del carburante cardiaco nei modelli murini provocano cardiomiopatia (Carley et al., 2014; Neubauer, 2007). In condizioni fisiologiche i cuori normali ossidano i corpi chetonici in proporzione alla loro consegna, a scapito dell'ossidazione degli acidi grassi e del glucosio, e il miocardio è il più alto consumatore di corpi chetonici per unità di massa (BING, 1954; Crawford et al., 2009; GARLAND et al. ., 1962; Hasselbaink et al., 2003; Jeffrey et al., 1995; Pelletier et al., 2007; Tardif et al., 2001; Yan et al., 2009). Rispetto all'ossidazione degli acidi grassi, i corpi chetonici sono più efficienti dal punto di vista energetico, producendo più energia disponibile per la sintesi di ATP per molecola di ossigeno investita (rapporto P / O) (Kashiwaya et al., 2010; Sato et al., 1995; Veech, 2004) . L'ossidazione del corpo chetonico produce anche energia potenzialmente più elevata della FAO, mantenendo l'ubichinone ossidato, il che aumenta la capacità redox nella catena di trasporto degli elettroni e rende disponibile più energia per sintetizzare l'ATP (Sato et al., 1995; Veech, 2004). L'ossidazione dei corpi chetonici può anche ridurre la produzione di ROS e quindi lo stress ossidativo (Veech, 2004).
Studi preliminari interventistici e osservazionali indicano un potenziale ruolo salutare dei corpi chetonici nel cuore. Nel contesto sperimentale dell'ischemia / riperfusione, i corpi chetonici hanno conferito potenziali effetti cardioprotettivi (Al-Zaid et al., 2007; Wang et al., 2008), probabilmente a causa dell'aumento dell'abbondanza mitocondriale nel cuore o dell'up-regolazione della fosforilazione ossidativa cruciale mediatori (Snorek et al., 2012; Zou et al., 2002). Studi recenti indicano che l'utilizzazione del corpo chetone è aumentata nel cuore di topi (Aubert et al., 2016) e nell'uomo (Bedi et al., 2016), supportando osservazioni precedenti nell'uomo (BING, 1954; Fukao et al., 2000; Janardhan et al., 2011; Longo et al., 2004; Rudolph e Schinz, 1973; Tildon e Cornblath, 1972). Le concentrazioni circolanti del corpo chetonico sono aumentate nei pazienti con insufficienza cardiaca, in proporzione diretta alle pressioni di riempimento, osservazioni il cui meccanismo e significato rimane sconosciuto (Kupari et al., 1995; Lommi et al., 1996; Lommi et al., 1997; Neely et al ., 1972), ma i topi con deficit di SCOT selettivo nei cardiomiociti mostrano un rimodellamento ventricolare patologico accelerato e firme ROS in risposta al danno da sovraccarico pressorio indotto chirurgicamente (Schugar et al., 2014).
Recenti osservazioni intriganti nella terapia del diabete hanno rivelato un potenziale legame tra il metabolismo chetone miocardico e il rimodellamento ventricolare patologico (Fig. 5). L'inibizione del co-trasportatore renale prossimale tubulare di sodio / glucosio 2 (SGLT2i) aumenta le concentrazioni circolanti di corpi chetonici negli esseri umani (Ferrannini et al., 2016a; Inagaki et al., 2015) e topi (Suzuki et al., 2014) aumentati chetogenesi epatica (Ferrannini et al., 2014; Ferrannini et al., 2016a; Katz e Leiter, 2015; Mudaliar et al., 2015). Sorprendentemente, almeno uno di questi agenti ha ridotto il ricovero in HF (ad esempio, come rivelato dallo studio EMPA-REG OUTCOME) e ha migliorato la mortalità cardiovascolare (Fitchett et al., 2016; Sonesson et al., 2016; Wu et al., 2016a ; Zinman et al., 2015). Mentre i meccanismi di guida dietro benefici esiti di HF a SGLT2i rimangono attivamente dibattuti, il beneficio di sopravvivenza è probabilmente multifattoriale, includendo in modo prospettico la chetosi ma anche effetti salutari su peso, pressione sanguigna, glucosio e livelli di acido urico, rigidità arteriosa, sistema nervoso simpatico, osmotico diuresi / riduzione del volume plasmatico e aumento dell'ematocrito (Raz e Cahn, 2016; Vallon e Thomson, 2016). Nel complesso, la nozione che aumentare la chetonemia terapeuticamente sia nei pazienti con scompenso cardiaco, sia in quelli ad alto rischio di sviluppare HF, rimane controversa ma è oggetto di studio attivo in studi pre-clinici e clinici (Ferrannini et al., 2016b; Kolwicz et al., 2016; Lopaschuk e Verma, 2016; Mudaliar et al., 2016; Taegtmeyer, 2016).

Corpi chetonici in biologia del cancro
I collegamenti tra corpi chetonici e cancro stanno rapidamente emergendo, ma gli studi su entrambi i modelli animali e sull'uomo hanno portato a conclusioni diverse. Poiché il metabolismo chetone è dinamico e reattivo allo stato nutritivo, è interessante per perseguire legami biologici con il cancro a causa del potenziale di terapie nutrizionali guidate con precisione. Le cellule tumorali sono sottoposte a riprogrammazione metabolica per mantenere una rapida proliferazione e crescita cellulare (DeNicola e Cantley, 2015, Pavlova e Thompson, 2016). Il classico effetto Warburg nel metabolismo delle cellule cancerogene deriva dal ruolo dominante della glicolisi e della fermentazione dell'acido lattico per trasferire energia e compensare una minore dipendenza dalla fosforilazione ossidativa e dalla limitata respirazione mitocondriale (De Feyter et al., 2016; Grabacka et al., 2016; Kang et al., 2015; Poff et al., 2014; Shukla et al., 2014). Il carbonio glucosio è principalmente diretto attraverso la glicolisi, la via del pentoso fosfato e la lipogenesi, che insieme forniscono gli intermedi necessari per l'espansione della biomassa tumorale (Grabacka et al., 2016; Shukla et al., 2014; Yoshii et al., 2015). L'adattamento delle cellule tumorali alla deprivazione di glucosio avviene attraverso la capacità di sfruttare fonti di carburante alternative, tra cui acetato, glutammina e aspartato (Jaworski et al., 2016; Sullivan et al., 2015). Ad esempio, l'accesso limitato al piruvato rivela la capacità delle cellule tumorali di convertire la glutammina in acetil-CoA mediante carbossilazione, mantenendo sia i bisogni energetici che anabolici (Yang et al., 2014). Un interessante adattamento delle cellule cancerose è l'utilizzo dell'acetato come combustibile (Comerford et al., 2014; Jaworski et al., 2016; Mashimo et al., 2014; Wright e Simone, 2016; Yoshii et al., 2015). L'acetato è anche un substrato per la lipogenesi, che è fondamentale per la proliferazione delle cellule tumorali, e il guadagno di questo condotto lipogenico è associato alla sopravvivenza dei pazienti più breve e al carico tumorale maggiore (Comerford et al., 2014; Mashimo et al., 2014; Yoshii et al ., 2015).
Le cellule non cancerose spostano facilmente la loro fonte di energia dal glucosio ai corpi chetonici durante la privazione del glucosio. Questa plasticità può essere più variabile tra i tipi di cellule tumorali, ma i tumori cerebrali impiantati in vivo hanno ossidato [2,4-13C2] -? OHB in misura simile al tessuto cerebrale circostante (De Feyter et al., 2016). I modelli con effetto Warburg inverso o metabolismo a due compartimenti ipotizzano che le cellule tumorali inducano la produzione di? OHB nei fibroblasti adiacenti, fornendo il fabbisogno energetico delle cellule tumorali (Bonuccelli et al., 2010; Martinez-Outschoorn et al., 2012) . Nel fegato, uno spostamento degli epatociti dalla chetogenesi all'ossidazione dei chetoni nelle cellule di carcinoma epatocellulare (epatoma) è coerente con l'attivazione delle attività BDH1 e SCOT osservate in due linee cellulari di epatoma (Zhang et al., 1989). In effetti, le cellule dell'epatoma esprimono OXCT1 e BDH1 e ossidano i chetoni, ma solo quando il siero muore di fame (Huang et al., 2016). In alternativa, è stata proposta anche la chetogenesi delle cellule tumorali. Cambiamenti dinamici nell'espressione genica chetogenica si manifestano durante la trasformazione cancerosa dell'epitelio del colon, un tipo di cellula che normalmente esprime HMGCS2, e un recente rapporto ha suggerito che HMGCS2 può essere un marker prognostico di prognosi sfavorevole nei carcinomi a cellule squamose e del colon-retto (Camarero et al., 2006; Chen et al., 2016). Resta da determinare se questa associazione richieda o coinvolga la chetogenesi o una funzione di luce lunare di HMGCS2. Al contrario, la produzione apparente di? OHB da parte delle cellule di melanoma e glioblastoma, stimolata dal PPAR? fenofibrato agonista, è stato associato all'arresto della crescita (Grabacka et al., 2016). Sono necessari ulteriori studi per caratterizzare i ruoli dell'espressione di HMGCS2 / SCOT, della chetogenesi e dell'ossidazione dei chetoni nelle cellule tumorali.
Al di là del regno del metabolismo del carburante, i chetoni sono stati recentemente coinvolti nella biologia delle cellule tumorali tramite un meccanismo di segnalazione. L'analisi del melanoma BRAF-V600E + ha indicato l'induzione di HMGCL dipendente da OCT1 in modo oncogenico BRAF-dipendente (Kang et al., 2015). L'aumento di HMGCL è stato correlato con una maggiore concentrazione di AcAc cellulare, che a sua volta ha migliorato l'interazione BRAFV600E-MEK1, amplificando la segnalazione MEK-ERK in un loop feed-forward che guida la proliferazione e la crescita delle cellule tumorali. Queste osservazioni sollevano l'interessante questione della chetogenesi extraepatica prospettica che quindi supporta un meccanismo di segnalazione (vedi anche? OHB come mediatore di segnalazione e Controversies in extraepatic chetogenesis). È anche importante considerare gli effetti indipendenti di AcAc, d-? OHB e l-? OHB sul metabolismo del cancro e, quando si considera l'HMGCL, anche il catabolismo della leucina può essere squilibrato.
Gli effetti delle diete chetogeniche (vedere anche Uso terapeutico della dieta chetogenica e dei corpi chetonici esogeni) nei modelli animali di cancro sono vari (De Feyter et al., 2016; Klement et al., 2016; Meidenbauer et al., 2015; Poff et al. ., 2014; Seyfried et al., 2011; Shukla et al., 2014). Mentre le associazioni epidemiologiche tra obesità, cancro e diete chetogeniche sono dibattute (Liskiewicz et al., 2016; Wright e Simone, 2016), una meta-analisi che utilizza diete chetogeniche in modelli animali e in studi sull'uomo ha suggerito un impatto salutare sulla sopravvivenza, con benefici prospetticamente collegati all'entità della chetosi, al momento dell'inizio della dieta e alla localizzazione del tumore (Klement et al., 2016; Woolf et al., 2016). Il trattamento delle cellule tumorali del pancreas con corpi chetonici (d-? OHB o AcAc) ha inibito la crescita, la proliferazione e la glicolisi e una dieta chetogenica (81% kcal di grassi, 18% di proteine, 1% di carboidrati) ha ridotto in vivo il peso del tumore, la glicemia e aumento del peso muscolare e corporeo negli animali con cancro impiantato (Shukla et al., 2014). Risultati simili sono stati osservati utilizzando un modello di cellule di glioblastoma metastatico in topi che hanno ricevuto l'integrazione di chetoni nella dieta (Poff et al., 2014). Al contrario, una dieta chetogenica (91% kcal di grassi, 9% di proteine) aumenta la concentrazione di OHB circolante e diminuisce la glicemia, ma non ha avuto alcun impatto sul volume del tumore o sulla durata di sopravvivenza nei ratti portatori di glioma (De Feyter et al., 2016). Un indice di glucosio chetone è stato proposto come indicatore clinico che migliora la gestione metabolica della terapia del cancro al cervello indotta dalla dieta chetogenica nell'uomo e nei topi (Meidenbauer et al., 2015). Presi insieme, i ruoli del metabolismo dei corpi chetonici e dei corpi chetonici nella biologia del cancro sono allettanti perché ciascuno di essi pone opzioni terapeutiche trattabili, ma gli aspetti fondamentali rimangono da chiarire, con chiare influenze che emergono da una matrice di variabili, comprese (i) differenze tra chetoni esogeni corpi contro dieta chetogenica, (ii) tipo di cellula cancerosa, polimorfismi genomici, grado e stadio; e (iii) tempi e durata dell'esposizione allo stato chetotico.

La chetogenesi è creata da corpi chetonici attraverso la scissione di acidi grassi e amminoacidi chetogenici. Questo processo biochimico fornisce energia a vari organi, in particolare il cervello, in circostanze di digiuno come risposta ad una non disponibilità di glucosio nel sangue. I corpi chetonici sono prodotti principalmente nei mitocondri delle cellule del fegato. Mentre altre cellule sono in grado di effettuare la chetogenesi, non sono così efficaci nel farlo come le cellule del fegato. Poiché la chetogenesi si verifica nei mitocondri, i suoi processi sono regolati indipendentemente. Dr. Alex Jimenez DC, CCST Insight
Applicazione terapeutica della dieta chetogenica e dei corpi chetonici esogeni
Le applicazioni delle diete chetogeniche e dei corpi chetonici come strumenti terapeutici sono sorte anche in contesti non cancerosi tra cui l'obesità e NAFLD / NASH (Browning et al., 2011; Foster et al., 2010; Schugar e Crawford, 2012); insufficienza cardiaca (Huynh, 2016; Kolwicz et al., 2016; Taegtmeyer, 2016); malattie neurologiche e neurodegenerative (Martin et al., 2016; McNally e Hartman, 2012; Rho, 2015; Rogawski et al., 2016; Yang e Cheng, 2010; Yao et al., 2011); errori congeniti del metabolismo (Scholl-B rgi et al, 2015); e la prestazione fisica (Cox et al., 2016). L'efficacia delle diete chetogeniche è stata particolarmente apprezzata nella terapia delle crisi epilettiche, in particolare nei pazienti resistenti ai farmaci. La maggior parte degli studi ha valutato le diete chetogeniche nei pazienti pediatrici e rivela una riduzione fino al 50% circa della frequenza delle crisi dopo 3 mesi, con una migliore efficacia in sindromi selezionate (Wu et al., 2016b). L'esperienza è più limitata nell'epilessia degli adulti, ma una riduzione simile è evidente, con una migliore risposta nei pazienti con epilessia generalizzata sintomatica (Nei et al., 2014). I meccanismi anticonvulsivanti sottostanti rimangono poco chiari, sebbene le ipotesi postulate includano un ridotto utilizzo / glicolisi del glucosio, trasporto riprogrammato del glutammato, impatto indiretto sul canale del potassio ATP-sensibile o sul recettore dell'adenosina A1, alterazione dell'espressione dell'isoforma del canale del sodio o effetti sugli ormoni circolanti inclusa la leptina ( Lambrechts et al., 2016; Lin et al., 2017; Lutas e Yellen, 2013). Non è chiaro se l'effetto anticonvulsivante sia principalmente attribuibile ai corpi chetonici o alle conseguenze metaboliche a cascata delle diete a basso contenuto di carboidrati. Tuttavia, gli esteri chetonici (vedi sotto) sembrano elevare la soglia convulsiva nei modelli animali di convulsioni provocate (Ciarlone et al., 2016; D'Agostino et al., 2013; Viggiano et al., 2015).
Le diete atkins e chetogeniche, a basso contenuto di carboidrati sono spesso considerate sgradevoli e possono causare stitichezza, iperuricemia, ipocalcemia, ipomagnesiemia, portare a nefrolitiasi, chetoacidosi, causare iperglicemia e aumentare il colesterolo circolante e le concentrazioni di acidi grassi liberi (Bisschop et al., 2001 ; Kossoff e Hartman, 2012; Kwiterovich et al., 2003; Suzuki et al., 2002). Per questi motivi, l'aderenza a lungo termine pone delle sfide. Gli studi sui roditori usano comunemente una distribuzione distintiva dei macronutrienti (94% kcal di grasso, 1% di carboidrati kcal, 5% di kcal di proteine, Bio-Serv F3666), che provoca una chetosi robusta. Tuttavia, l'aumento del contenuto proteico, anche a 10% kcal, diminuisce sostanzialmente la chetosi e la restrizione della proteina 5% kcal conferisce confondenti effetti metabolici e fisiologici. Questa formulazione dietetica è anche colina esaurita, un'altra variabile che influenza la suscettibilità al danno epatico e persino la chetogenesi (Garbow et al., 2011; Jornayvaz et al., 2010; Kennedy et al., 2007; Pissios et al., 2013; Schugar et al., 2013). Gli effetti del consumo a lungo termine di diete chetogeniche nei topi rimangono incompleti, ma studi recenti sui topi hanno rivelato una sopravvivenza normale e l'assenza di marcatori di danno epatico nei topi su diete chetogeniche nel corso della loro vita, sebbene il metabolismo degli aminoacidi, il dispendio energetico e la segnalazione di insulina sono stati notevolmente riprogrammati (Douris et al., 2015).
Meccanismi che aumentano la chetosi attraverso meccanismi alternativi alle diete chetogeniche includono l'uso di precursori del corpo chetone ingestibili. La somministrazione di corpi chetonici esogeni potrebbe creare uno stato fisiologico unico non riscontrato nella fisiologia normale, poiché le concentrazioni di glucosio e insulina circolanti sono relativamente normali, mentre le cellule potrebbero risparmiare l'assorbimento e l'utilizzo di glucosio. Gli stessi corpi chetonici hanno una breve emivita e l'ingestione o l'infusione di sale di sodio-OHB per ottenere la chetosi terapeutica provoca un carico di sodio indesiderato. L'R / S-1,3-butandiolo è un dialcool non tossico che viene facilmente ossidato nel fegato per produrre d / l-? OHB (Desrochers et al., 1992). In contesti sperimentali distinti, questa dose è stata somministrata quotidianamente a topi o ratti per un massimo di sette settimane, producendo concentrazioni di? OHB circolanti fino a 5 mM entro 2 ore dalla somministrazione, che è stabile per almeno altre 3 ore (D ' Agostino et al., 2013). La soppressione parziale dell'assunzione di cibo è stata osservata nei roditori dati R / S-1,3-butandiolo (Carpenter and Grossman, 1983). Inoltre, tre esteri chetonici (KE) chimicamente distinti, (i) monoestere di R-1,3-butandiolo e d-? OHB (R-3-idrossibutil R-? OHB); (ii) gliceril-tris-? OHB; e (iii) R, S-1,3-butandiolo acetoacetato diestere, sono stati anche ampiamente studiati (Brunengraber, 1997; Clarke et al., 2012a; Clarke et al., 2012b; Desrochers et al., 1995a; Desrochers et al. ., 1995b; Kashiwaya et al., 2010). Un vantaggio intrinseco del primo è che 2 moli di d-? OHB fisiologico vengono prodotte per mole di KE, in seguito all'idrolisi dell'esterasi nell'intestino o nel fegato. La sicurezza, la farmacocinetica e la tolleranza sono state studiate più ampiamente negli esseri umani che ingeriscono R-3-idrossibutil R-? OHB, a dosi fino a 714 mg / kg, producendo concentrazioni circolanti di d-? OHB fino a 6 mM (Clarke et al., 2012a; Cox et al., 2016; Kemper et al., 2015; Shivva et al., 2016). Nei roditori, questo KE riduce l'apporto calorico e il colesterolo totale plasmatico, stimola il tessuto adiposo bruno e migliora la resistenza all'insulina (Kashiwaya et al., 2010; Kemper et al., 2015; Veech, 2013). Recenti scoperte indicano che durante l'esercizio in atleti allenati, l'ingestione di R-3-idrossibutil R-? OHB ha diminuito la glicolisi dei muscoli scheletrici e le concentrazioni di lattato plasmatico, aumentato l'ossidazione intramuscolare del triacilglicerolo e conservato il contenuto di glicogeno muscolare, anche quando i carboidrati ingeriti hanno stimolato la secrezione di insulina ( Cox et al., 2016). È necessario un ulteriore sviluppo di questi risultati intriganti, poiché il miglioramento delle prestazioni degli esercizi di resistenza è stato determinato principalmente da una robusta risposta al KE nei soggetti 2 / 8. Tuttavia, questi risultati supportano studi classici che indicano una preferenza per l'ossidazione chetone su altri substrati (GARLAND et al., 1962; Hasselbaink et al., 2003; Stanley et al., 2003; Valente-Silva et al., 2015), incluso durante l'esercizio, e che gli atleti addestrati possono essere più predisposti ad utilizzare i chetoni (Johnson et al., 1969a; Johnson e Walton, 1972; Winder et al., 1974; Winder et al., 1975). Infine, restano da determinare i meccanismi che potrebbero supportare una migliore prestazione fisica a parità di apporto calorico (differenzialmente distribuito tra macronutrienti) e tassi di consumo di ossigeno uguali.
Prospettiva futura
Una volta ampiamente stigmatizzata come una via di trabocco in grado di accumulare emissioni tossiche dalla combustione dei grassi in stati di ristrettezze di carboidrati (il paradigma `` chetotossico ''), recenti osservazioni supportano l'idea che il metabolismo corporeo chetone svolga ruoli salutari anche negli stati carichi di carboidrati, aprendo una `` chetoormetica '' ipotesi. Mentre i facili approcci nutrizionali e farmacologici per manipolare il metabolismo dei chetoni lo rendono un bersaglio terapeutico attraente, gli esperimenti proposti in modo aggressivo ma prudenti rimangono sia nei laboratori di ricerca di base che in quelli traslazionali. Sono emerse esigenze insoddisfatte nell'ambito della definizione del ruolo di sfruttare il metabolismo dei chetoni nell'insufficienza cardiaca, nell'obesità, nella NAFLD / NASH, nel diabete di tipo 2 e nel cancro. La portata e l'impatto dei ruoli di segnalazione `` non canonici '' dei corpi chetonici, inclusa la regolazione dei PTM che probabilmente si alimentano avanti e indietro nelle vie metaboliche e di segnalazione, richiedono un'esplorazione più approfondita. Infine, la chetogenesi extraepatica potrebbe aprire intriganti meccanismi di segnalazione paracrina e autocrina e opportunità per influenzare il co-metabolismo all'interno del sistema nervoso e dei tumori per raggiungere scopi terapeutici.
Ringraziamenti
Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/
Le note
In conclusione, i corpi chetonici vengono creati dal fegato per essere utilizzati come fonte di energia quando non c'è abbastanza glucosio prontamente disponibile nel corpo umano. La chetogenesi si verifica quando ci sono bassi livelli di glucosio nel sangue, in particolare dopo che le altre riserve di carboidrati cellulari sono state esaurite. Lo scopo dell'articolo sopra era quello di discutere i ruoli multidimensionali dei corpi chetonici nel metabolismo del carburante, nella segnalazione e nella terapia. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
A cura di Dr. Alex Jimenez
Riferito da: Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/

Discussione aggiuntiva sull'argomento: Dolore alla schiena acuto
Mal di schienaè una delle cause più diffuse di disabilità e di giornate di lavoro perse in tutto il mondo. Il dolore alla schiena si attribuisce al secondo motivo più comune per le visite presso l'ambulatorio medico, superato solo dalle infezioni delle vie respiratorie superiori. Circa l'80% della popolazione sperimenterà dolore alla schiena almeno una volta nella vita. La colonna vertebrale è una struttura complessa composta da ossa, articolazioni, legamenti e muscoli, tra gli altri tessuti molli. Lesioni e / o condizioni aggravate, come dischi erniciati, può eventualmente portare a sintomi di mal di schiena. Gli infortuni sportivi o gli incidenti automobilistici sono spesso la causa più frequente di mal di schiena, tuttavia, a volte il più semplice dei movimenti può avere risultati dolorosi. Fortunatamente, opzioni di trattamento alternative, come la cura chiropratica, possono aiutare ad alleviare il mal di schiena attraverso l'uso di aggiustamenti spinali e manipolazioni manuali, migliorando in definitiva il sollievo dal dolore.

EXTRA EXTRA | ARGOMENTO IMPORTANTE: consigliato El Paso, TX Chiropractor
***