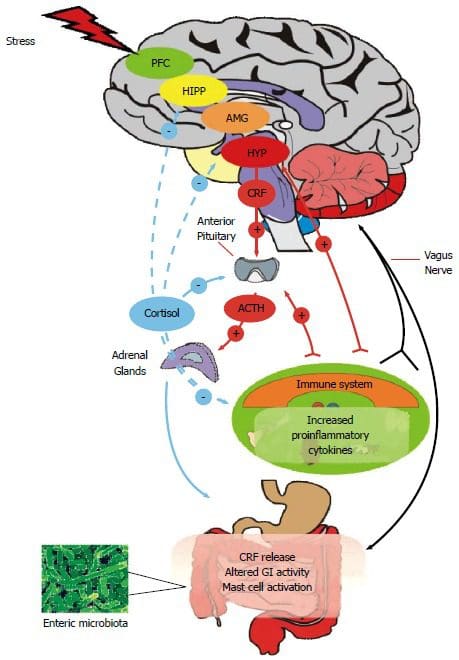
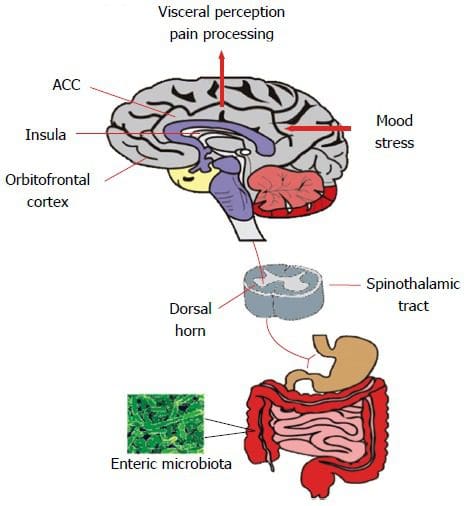
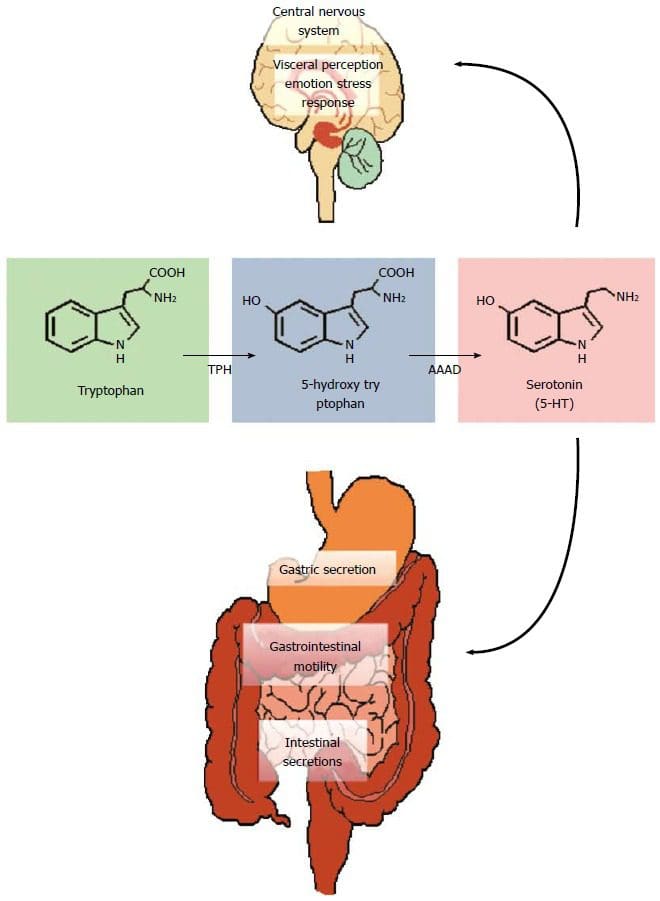
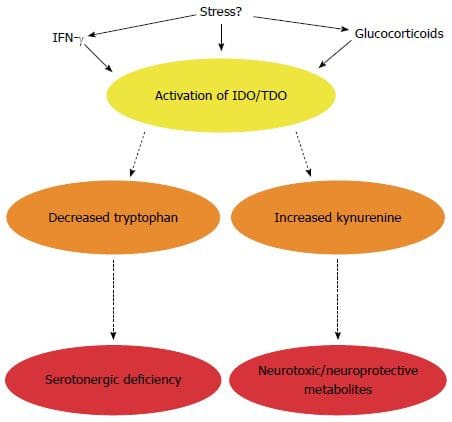
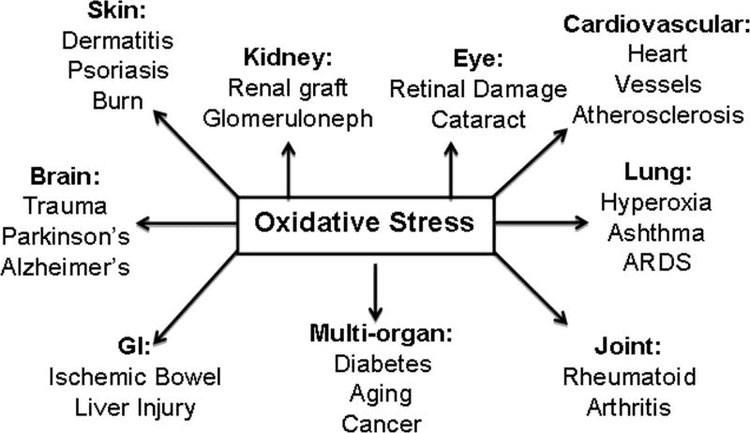
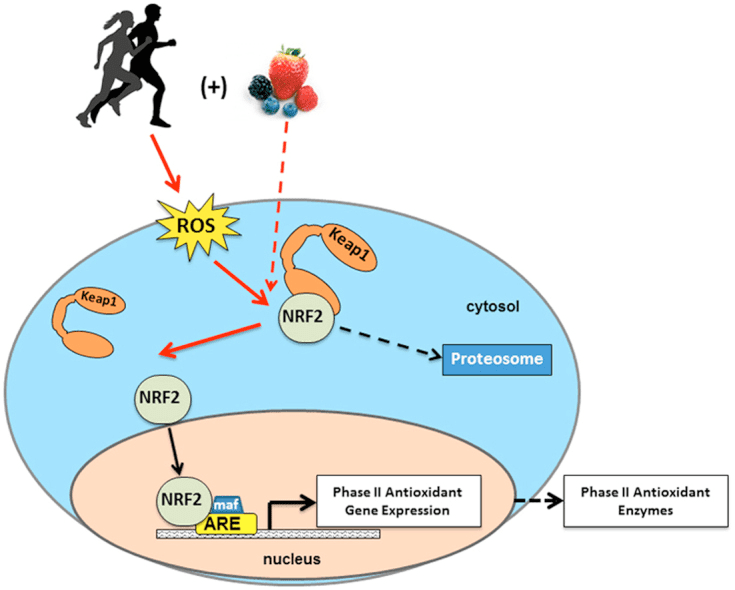
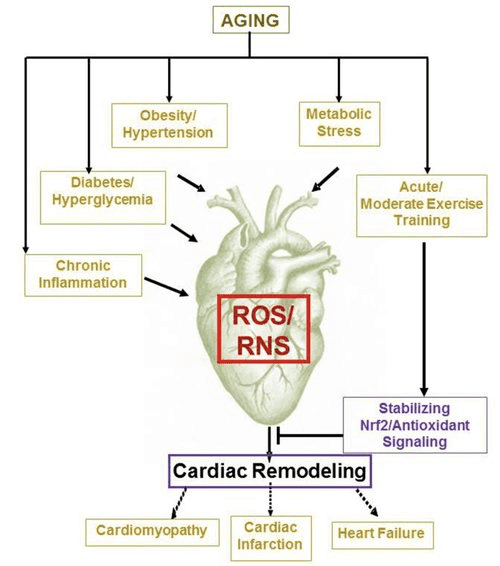
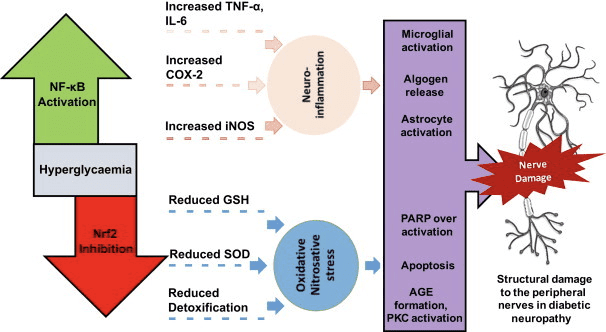
Back Clinic Team di Chiropratica e Medicina Funzionale Stress Ossidativo. Lo stress ossidativo è definito come un disturbo nell'equilibrio tra la produzione di ossigeno reattivo (radicali liberi) e le difese antiossidanti. In altre parole, si tratta di uno squilibrio tra la produzione di radicali liberi e la capacità dell'organismo di contrastare o disintossicare gli effetti nocivi attraverso la neutralizzazione da parte degli antiossidanti. Lo stress ossidativo porta a molte condizioni fisiopatologiche nel corpo. Questi includono malattie neurodegenerative, ad esempio morbo di Parkinson, morbo di Alzheimer, mutazioni genetiche, tumori, sindrome da stanchezza cronica, sindrome dell'X fragile, disturbi cardiaci e dei vasi sanguigni, aterosclerosi, insufficienza cardiaca, infarto e malattie infiammatorie. L'ossidazione avviene in una serie di circostanze:
le cellule usano il glucosio per produrre energia
il sistema immunitario sta combattendo contro i batteri e creando infiammazioni
i corpi detossificano inquinanti, pesticidi e fumo di sigaretta
Ci sono milioni di processi che avvengono nei nostri corpi in un dato momento che possono provocare l'ossidazione. Ecco alcuni sintomi:
stanchezza
Perdita di memoria e / o nebbia cerebrale
Dolore muscolare e articolare
Rughe e capelli grigi
Diminuzione della vista
Mal di testa e sensibilità al rumore
Suscettibilità alle infezioni
Scegliere cibi biologici ed evitare le tossine nel proprio ambiente fa una grande differenza. Questo, insieme alla riduzione dello stress, può essere utile per ridurre l'ossidazione.
Svegliarsi stanco anche dopo aver dormito sei o più ore di sonno?
Sotto un alto livello di stress?
Se si verifica una di queste situazioni, ciò potrebbe essere dovuto ai livelli di melatonina e cortisolo che influenzano il corpo e il ritmo circadiano.
In tutto il mondo, milioni di persone hanno difficoltà a dormire. Negli Stati Uniti ce ne sono circa 50-70 milioni di persone che hanno una scarsa qualità del sonno. Quando una persona ha dormito per meno di otto ore, si stanca e possono presentarsi molti problemi, soprattutto se la sua vita è frenetica. Con uno stile di vita frenetico e un sonno scarso, il corpo può avere una bassa energia per svolgere qualsiasi compito, l'ormone dello stress cortisolo verrà sollevato e malattie come l'ipertensione e il diabete possono causare problemi che possono essere cronici se non lo sono trattata.
Nell'endocrinologia funzionale, la melatonina e il cortisolo sono ormoni che il corpo produce naturalmente. L'ormone cortisolo o l'ormone dello stress aiuta il corpo ad essere in uno stato di "lotta o fuga", che può essere una buona cosa per chiunque stia facendo un progetto o andando a un colloquio di lavoro. Anche se quando i livelli di ormone cortisolo sono alti, può portare al corpo ad avere complicazioni come infiammazione, stress ossidativo cronico e ipertensione.
Il ritmo circadiano della melatonina
Con l'ormone melatonina, questo ormone dice al corpo quando è il momento di dormire. A volte, tuttavia, le persone hanno difficoltà a dormire e l'assunzione di integratori di melatonina può effettivamente rilassare il corpo e far addormentare la persona. Poiché la ghiandola pineale produce melatonina dal cervello, può anche essere trovata negli occhi, nel midollo osseo e nell'intestino per rilassare il corpo e far addormentare naturalmente la persona. Alcuni studi dimostrano che il ritmo circadiano della ghiandola pineale che produce melatonina. In questo modo, la ricerca mostra che la somministrazione di melatonina può:
Uno: indurre il sonno su individui che hanno difficoltà ad addormentarsi.
Due: impedisce al corpo di svegliarsi naturalmente dal pacemaker circadiano.
Tre: sposta gli orologi biologici circadiani per aumentare l'assunzione di sonno quando una persona sta cercando di dormire in un momento precedente per ottenere tutti i benefici di otto ore del sonno.
Quando una persona lavora con un lavoro dalle 9 alle 5, si alza con il proprio corpo e rilassa il proprio corpo dopo una dura giornata di lavoro. Gli studi hanno scoperto che gli ormoni della melatonina e del cortisolo aiutano a regolare il modello di 24 ore della funzione del corpo e le risposte tremendamente. Con il ciclo di produzione dell'ormone del corpo, può essere disturbato se la persona sta sveglia a tarda notte o dorme durante il giorno. Quando ciò accade, la persona può avere disturbi distruttivi come sbalzi d'umore, vertigini, essere irritabile e depresso e avere disturbi metabolici. Non solo, ma anche il sistema immunitario e il suo sistema endocrino possono essere danneggiati, causando l'organismo ospite di infezioni e malattie.
Ci sono stati più studi sui ritmi circadiani nel corpo, come gli studi mostrano come le persone che lavorano nel turno di notte sono state associate a un gran numero di problemi di salute avversi che attaccano il sistema cardiovascolare e gastrointestinale e disturbano il sistema metabolico. Chiunque abbia lavorato il turno di notte deve cambiare il proprio programma di sonno e adattarsi al rapido riorientamento del proprio programma di sonno / veglia per andare a lavorare e fare il proprio lavoro. Dal momento che tutti lavorano a turni, può essere stressante e influire sulle prestazioni corporee di un lavoratore, nonché sulla secrezione di melatonina e cortisolo.
Modi per supportare il cortisolo e la melatonina
Sorprendentemente, ci sono modi per abbassare i livelli di cortisolo e assicurarsi che i livelli di melatonina funzionino correttamente affinché il corpo funzioni. Per abbassare i livelli di cortisolo, una persona dovrebbe praticare pratiche meditative, trovare un passatempo divertente e, soprattutto, provare esercizi di respirazione profonda per rilassare il corpo dallo stress indesiderato. Con esercizi di respirazione profonda, può aiutare il corpo a rilasciare qualsiasi tensione che una persona tiene, ei muscoli del corpo hanno iniziato a rilassarsi e il sangue inizia a fluire. Con i livelli di melatonina, lavorano insieme al ritmo circadiano del corpo e si assicurano che il corpo sappia quando è il momento di svegliarsi, dormire e mangiare. L'ormone melatonina può anche aiutare a regolare la temperatura corporea, pressione sanguignae livelli ormonali per assicurarsi che funzioni correttamente. Quando ci sono alti livelli di questi sistemi, può causare il corpo a sviluppare malattie croniche e danneggiare il corpo nel processo.
La ricerca mostra che gli ormoni della melatonina possono legarsi ai recettori neurologici nel corpo, promuovendo così il rilassamento. Poiché la melatonina si lega ai recettori neurologici, può anche ridurre l'attività dei nervi e i livelli di dopamina per rendere gli occhi pesanti, facendo addormentare la persona.
Conclusione
Con il corpo in grado di produrre naturalmente i livelli di melatonina e cortisolo per assicurarsi che il corpo non sia eccessivamente stressato durante tutto il giorno. Poiché la melatonina è associata al ritmo circadiano del corpo, il corpo sa quando stare in piedi e addormentarsi. Poiché tutti hanno un programma frenetico, è essenziale prendere tempo e rilassarsi e seguire un programma di sonno sano in modo che il corpo possa essere sano e funzionante. Alcuni prodotti sono qui per assicurarsi che il sistema endocrino funzioni correttamente e sostenga le ghiandole surrenali e il metabolismo dello zucchero.
Lo scopo delle nostre informazioni è limitato a problemi di chiropratica, muscoloscheletrici e di salute nervosa o articoli, argomenti e discussioni di medicina funzionale. Utilizziamo protocolli sanitari funzionali per il trattamento di lesioni o disturbi del sistema muscoloscheletrico. Il nostro ufficio ha fatto un ragionevole tentativo di fornire citazioni di supporto e ha identificato gli studi di ricerca pertinenti o studi a supporto dei nostri posti. Facciamo anche copie degli studi di ricerca di supporto disponibili al consiglio e / o al pubblico su richiesta. Per discutere ulteriormente l'argomento di cui sopra, non esitate a chiedere al Dr. Alex Jimenez o contattarci al numero 915-850-0900.
Riferimenti:
Cajochen, C, et al. Ruolo della melatonina nella regolazione dei ritmi circadiani umani e del sonno . Journal of Neuroendocrinology, Biblioteca nazionale americana di medicina, aprile 2003, www.ncbi.nlm.nih.gov/pubmed/12622846.
James, Francine O, et al. Ritmi circadiani di melatonina, cortisolo e espressione genica dell'orologio durante il lavoro notturno simulato. Pernottamento, Associated Professional Sleep Societies, LLC, novembre 2007, www.ncbi.nlm.nih.gov/pmc/articles/PMC2082093/.
Monteleone, P, et al. Rapporto temporale tra le risposte della melatonina e del cortisolo allo stress fisico notturno negli esseri umani . Psychoneuroendocrinology, Biblioteca nazionale americana di medicina, 1992, www.ncbi.nlm.nih.gov/pubmed/1609019.
Raman, Ryan. "In che modo la melatonina può aiutarti a dormire e sentirti meglio" Healthline, Healthline Media, 3 settembre 2017, www.healthline.com/nutrition/melatonin-and-sleep.
Zamanian, Zahra, et al. Outline of Changes in Cortisol and Melatonin Circadian Rhythms in the Security Guards of Shiraz University of Medical Sciences. Giornale internazionale di medicina preventiva, Medknow Publications & Media Pvt Ltd, luglio 2013, www.ncbi.nlm.nih.gov/pmc/articles/PMC3775223/.
Informando le persone su come l'Università Nazionale di Scienze della Salute fornisce le conoscenze per le generazioni future che vogliono fare la differenza nel mondo. L'Università offre una vasta gamma di professioni mediche per la medicina funzionale e integrativa.
Ti sei mai chiesto perché ti senti pigro dopo una lunga giornata? O ti senti male allo stomaco quando hai mangiato qualcosa di cattivo o ti sei lasciato andare al tuo cibo preferito? Potrebbe essere che il tuo intestino mostri segni di stress e disagio a causa di certe abitudini che potresti incontrare e non lo sapevi nemmeno?
Nel nostro precedente articolo, ne abbiamo parlato i sei tipi di cibo che il nostro intestino deve essere sano. Dal nostro istinto contiene trilioni di microbiomi, sia buoni che cattivi, questi microbiomi svolgono un ruolo importante nella nostra salute generale. Un microbioma sano migliora il nostro salute dello stomaco, salute del cuore, salute del cervello, controlla il nostro peso ed regola il nostro zucchero nel sangue. Con i batteri buoni nel nostro intestino, i batteri ci avvantaggiano con un buon sistema digestivo e distruggono i batteri dannosi. Ma alcuni stili di vita e scelte dietetiche possono effettivamente aumentare i batteri cattivi e abbassare i batteri buoni e la salute generale.
Ecco cinque scelte sorprendentemente di stile di vita che ti fanno male all'intestino:
Non mangiare una vasta gamma di alimenti
Il nostro intestino svolge un ruolo importante nella nostra salute generale. Quando mangiamo buoni cibi integrali, il nostro intestino è più felice; abbiamo più energia per completare qualsiasi compito che ci viene lanciato e stiamo ottenendo nutrienti per la nostra flora intestinale. Tuttavia, negli ultimi due decenni, ci siamo concentrati maggiormente sugli alimenti trasformati a causa delle pressioni economiche dell'aumento delle produzioni alimentari. FOA ha affermato che "il 75% del cibo del mondo è generato da solo 12 piante e cinque specie animali" e questo è molto dannoso per la nostra flora intestinale.
Qui a Injury Medical & Chiropractic Clinic, informiamo i nostri pazienti sull'importanza di mangiare cibi integrali e nutrienti per promuovere non solo un intestino sano ma una mente sana. Quando il corpo viene presentato a un file ampia varietà di cibi integrali (con un alto contenuto di fibre), il nostro intestino inizia a riparare i danni degli alimenti trasformati che potremmo aver consumato internamente.
Tuttavia, quando trascuri i prebiotici nella tua dieta, lo sei danneggiare la salute dell'apparato digerente. Senza prebiotici, il nostro sistema digestivo rallenta lo sviluppo e la diversità della nostra flora intestinale. Quindi per avere un microbioma sano sviluppo, è necessario incorporare alimenti pieni di fibre digeribili e indigeribili nella dieta. Alcuni alimenti inclusi in questa categoria sono avena, noci, cipolle, aglio, porri, asparagi, banane, pere, ceci e fagioli.
Attenersi a una dieta ricca di fibre potrebbe essere impegnativo, tuttavia c'è la possibilità di assumere integratori prebiotici. Se hai un allergene alimentare o sensibilità alimentare a qualsiasi cibo ricco di fibre, prendi integratori prebiotici può effettivamente aiutare a far crescere Bifidobacterium e Faecalibacterium nell'intestino ed essere benefico per la salute senza il disagio.
Eccessivo consumo di alcol
Ogni adulto gode di alcol di tanto in tanto. Sì, è una di quelle bevande che ti aiutano a rilassarti un po 'dopo una lunga giornata, tuttavia troppo può portare ad abuso di alcol e dipendenza. Quindi, lo sapevi che consumare così tanto alcol è dannoso il tuo cuore, fegato e cervello; ferendo così la salute dell'intestino e dandoti la disbiosi?
Uno studio ha affermato che gli alcolisti con disbiosi avevano un'abbondanza mediana inferiore di Bacteroidetes e un'abbondanza elevata di Proteobacteria. Quelli che non erano alcolisti non sono stati influenzati dallo studio.
Però; ci sono alcune buone notizie su come limitarsi all'alcolismo e che può essere benefico per i batteri intestinali. Se hai consumato moderatamente vino rosso in modo responsabile, il polifenoli nel vino può aiutare la tua flora intestinale. Quindi, goditi un bicchiere di vino di tanto in tanto come una piccola sorpresa che non dovrebbe essere dato per scontato.
Sonno inadeguato
In uno degli articoli precedenti, abbiamo parlato di come ottenere un buona notte di sonno attraverso le erbe. Quando dormiamo poco o niente durante la nostra vita frenetica, ci colpisce attraverso vari problemi di salute, tra cui malattia cardiaca ed obesità. In un uno studio del 2016 , i ricercatori hanno scoperto l'effetto della privazione del sonno a breve termine sul microbiota intestinale dopo due giorni.
Quando il nostro corpo non riceve le 8 ore di sonno raccomandate, il nostro intestino subisce un enorme tributo perché ci sentiamo pigri ed esausti. Quindi, per assicurarci che il nostro microbioma intestinale sarà curato, ti consigliamo di spegnere i tuoi dispositivi elettronici almeno 30 minuti prima di prepararti a sistemarti per la notte. Spegni tutte le luci e non bere liquidi almeno due ore prima di andare a letto, chiudi gli occhi e fai un respiro profondo in uno stato meditativo e rilassati mentre ti addormenti nella città del sonno.
Esercizio inadeguato
Attraverso il nostro stile di vita frenetico e il lavoro stressante, è difficile trovare il tempo per fare esercizio. Ma quando troviamo effettivamente il tempo per fare esercizio, non solo la nostra mente si sente bene; ma anche il nostro corpo e il nostro intestino si sentono bene. Tuttavia, le cose vengono sempre fuori quando siamo in una routine di esercizi e dobbiamo saltare del tutto l'esercizio. Succede a tutti noi ed è difficile riprendere da dove avevamo interrotto quando abbiamo provato a fare esercizio.
Quando non ci esercitiamo almeno un paio di volte alla settimana, il nostro corpo ci fa un sacco di soldi man mano che aumentiamo di peso, il nostro lo stress è troppo altoe abbiamo un maggiori possibilità di avere una malattia cronica. Quando ciò accade, la nostra flora intestinale è un enorme svantaggio. Qui alla clinica, ci sforziamo di informare i nostri pazienti sull'importanza dell'esercizio e che non solo cambia la loro vita ma cambia anche il loro umore.
Tuttavia, non limitarti a un duro esercizio di routine in cui ti ferirai. Inizia con un allenamento a bassa intensità, quindi aumentalo man mano che la tua flora intestinale ti ringrazierà per questo.
Come ultima affermazione, noi di Injury Medical vogliamo tenervi informati sulla nutrizione e sui modi per aiutarvi a migliorare i vostri disturbi con queste sorprese 5. Ma anche per educarti su ciò che potrebbe farti male all'intestino. Con queste sorprese e lievi cambiamenti nella tua vita quotidiana, il tuo istinto ti ringrazierà per il lungo raggio.
Risorse NCBI
Secondo le prove di uno studio di ricerca 2016, il sistema immunitario dell'intestino è fondamentale per prevenire una varietà di malattie e spesso può contribuire a disturbi metabolici. Tuttavia, potrebbe anche aiutare a fornire un obiettivo di trattamento quando si osserva l'infiammazione sistemica nella resistenza all'insulina. Inoltre, l'immunità intestinale modificata è stata collegata a cambiamenti nel microbiota intestinale, funzione di barriera intestinale, cellule immunitarie intestinali e resistenza agli antigeni che entrano nel sistema gastrointestinale o gastrointestinale. Sebbene in precedenza si credesse che ciò aumentasse il pericolo di disturbi esofagei tra cui infezioni patogene e infiammazioni croniche, che alla fine possono portare a problemi di salute cronici.
I corpi chetonici sono creati dal fegato e utilizzati come fonte di energia quando il glucosio non è prontamente disponibile nel corpo umano. I due corpi chetonici principali sono l'acetoacetato (AcAc) e il 3-beta-idrossibutirrato (3HB), mentre l'acetone è il terzo e meno abbondante corpo chetonico. I chetoni sono sempre presenti nel sangue e il loro livello aumenta durante il digiuno e l'esercizio prolungatochetogenesi è il processo biochimico mediante il quale gli organismi producono corpi chetonici attraverso la scissione di acidi grassi e amminoacidi chetogenici.
I corpi chetonici sono generati principalmente nel mitocondri di cellule del fegato. La chetogenesi si verifica quando vi sono bassi livelli di glucosio nel sangue, in particolare dopo che sono state esaurite altre riserve di carboidrati cellulari, come il glicogeno. Questo meccanismo può anche verificarsi quando non vi sono quantità sufficienti di insulina. La produzione di corpi chetonici viene infine avviata per rendere disponibile l'energia immagazzinata nel corpo umano sotto forma di acidi grassi. La chetogenesi si verifica nei mitocondri dove è regolata indipendentemente.
Astratto
Il metabolismo del corpo chetone è un nodo centrale nell'omeostasi fisiologica. In questa recensione, discutiamo di come i chetoni servono ruoli metabolici discreti a regolazione fine che ottimizzano le prestazioni di organi e organismi in vari resti di nutrienti e proteggono da infiammazioni e lesioni in sistemi di organi multipli. Tradizionalmente visti come substrati metabolici arruolati solo nella restrizione dei carboidrati, recenti osservazioni sottolineano l'importanza dei corpi chetonici come metabolici metabolici e di segnalazione vitali quando i carboidrati sono abbondanti. A complemento di un repertorio di opzioni terapeutiche conosciute per le malattie del sistema nervoso, sono emersi i ruoli potenziali per i corpi chetonici nel cancro, così come i ruoli protettivi intriganti nel cuore e nel fegato, aprendo le opzioni terapeutiche nelle malattie legate all'obesità e cardiovascolari. Le controversie nel metabolismo e segnalazione dei chetoni sono discusse per conciliare il dogma classico con le osservazioni contemporanee.
Introduzione
I corpi chetonici sono una fonte vitale alternativa di combustibile metabolico per tutti i domini della vita, eucarya, batteri e archei (Aneja et al., 2002; Cahill GF Jr, 2006; Krishnakumar et al., 2008). Il metabolismo del corpo chetonico negli esseri umani è stato sfruttato per alimentare il cervello durante periodi episodici di privazione dei nutrienti. I corpi chetonici sono intrecciati con le vie metaboliche cruciali dei mammiferi come l'ossidazione β (FAO), il ciclo degli acidi tricarbossilici (TCA), la gluconeogenesi, la lipogenesi de novo (DNL) e la biosintesi degli steroli. Nei mammiferi, i corpi chetonici sono prodotti prevalentemente nel fegato dall'acetil-CoA derivato dalla FAO e vengono trasportati nei tessuti extraepatici per l'ossidazione terminale. Questa fisiologia fornisce un carburante alternativo che è aumentato da periodi relativamente brevi di digiuno, che aumenta la disponibilità di acidi grassi e diminuisce la disponibilità di carboidrati (Cahill GF Jr, 2006; McGarry e Foster, 1980; Robinson e Williamson, 1980). L'ossidazione del corpo chetonico diventa un contributo significativo al metabolismo energetico complessivo dei mammiferi all'interno dei tessuti extraepatici in una miriade di stati fisiologici, tra cui digiuno, fame, periodo neonatale, post-esercizio, gravidanza e aderenza a diete a basso contenuto di carboidrati. Le concentrazioni corporee di chetoni totali circolanti in esseri umani adulti sani normalmente mostrano oscillazioni circadiane tra circa 100-250 M, salgono a ~ 1 mM dopo un esercizio prolungato o 24 ore di digiuno e possono accumularsi fino a 20 mM in stati patologici come la chetoacidosi diabetica ( Cahill GF Jr, 2006; Johnson et al., 1969b; Koeslag et al., 1980; Robinson e Williamson, 1980; Wildenhoff et al., 1974). Il fegato umano produce fino a 300 g di corpi chetonici al giorno (Balasse e Fery, 1989), che contribuiscono tra il 5 e il 20% del dispendio energetico totale negli stati nutriti, a digiuno e affamati (Balasse et al., 1978; Cox et al. al., 2016).
Recenti studi ora evidenziano ruoli imperativi per corpi chetonici nel metabolismo delle cellule dei mammiferi, omeostasi e segnalazione in un'ampia varietà di stati fisiologici e patologici. Oltre a servire come combustibile energetico per i tessuti extraepatici come cervello, cuore o muscolo scheletrico, i corpi chetonici svolgono ruoli chiave come mediatori segnalatori, fattori di modificazione della proteina post-traduzionale (PTM) e modulatori dell'infiammazione e dello stress ossidativo. In questa recensione, forniamo sia visioni classiche che moderne dei ruoli pleiotropici dei corpi chetonici e del loro metabolismo.
Panoramica del metabolismo del corpo chetone
Il tasso di chetogenesi epatica è regolato da una serie orchestrata di trasformazioni fisiologiche e biochimiche del grasso. I regolatori primari includono la lipolisi degli acidi grassi dai triacilgliceroli, il trasporto verso e attraverso la membrana plasmatica degli epatociti, il trasporto nei mitocondri tramite la carnitina palmitoiltransferasi 1 (CPT1), la spirale di α-ossidazione, l'attività del ciclo TCA e le concentrazioni intermedie, il potenziale redox e i regolatori ormonali di questi processi, prevalentemente glucagone e insulina [recensito in (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al., 1983; Kahn et al., 2005; McGarry e Foster , 1980; Williamson et al., 1969)]. Classicamente la chetogenesi è vista come una via di ricaduta, in cui l'acetil-CoA derivato dall'ossidazione α supera l'attività di citrato sintasi e / o la disponibilità di ossalacetato per la condensazione per formare citrato. Gli intermedi a tre atomi di carbonio mostrano attività anti-chetogenica, presumibilmente a causa della loro capacità di espandere il pool di ossalacetato per il consumo di acetil-CoA, ma la concentrazione di acetil-CoA epatica da sola non determina la velocità chetogenica (Foster, 1967; Rawat e Menahan, 1975; Williamson et al., 1969). La regolazione della chetogenesi da eventi ormonali, trascrizionali e post-traduzionali insieme supportano l'idea che i meccanismi molecolari che regolano la velocità chetogenica rimangono non completamente compresi (vedi Regolamento di HMGCS2 e SCOT / OXCT1).
La chetogenesi si verifica principalmente nella matrice mitocondriale epatica a velocità proporzionali all'ossidazione totale dei grassi. Dopo il trasporto delle catene aciliche attraverso le membrane mitocondriali e l'ossidazione a, l'isoforma mitocondriale della 3-idrossimetilglutaril-CoA sintasi (HMGCS2) catalizza il destino commettendo la condensazione dell'acetoacetil-CoA (AcAc-CoA) e dell'acetil-CoA per generare HMGCS1 (Fig. 1A). L'HMG-CoA liasi (HMGCL) scinde l'HMG-CoA per liberare acetil-CoA e acetoacetato (AcAc), e quest'ultimo viene ridotto a d -? - idrossibutirrato (d-? OHB) dalla d-? OHB deidrogenasi mitocondriale dipendente dalla fosfatidilcolina ( BDH1975) in una reazione di quasi equilibrio accoppiata a NAD + / NADH (Bock e Fleischer, 1960; LEHNINGER et al., 1). La costante di equilibrio BDH1 favorisce la produzione di d-? OHB, ma il rapporto tra corpi chetonici AcAc / d-? OHB è direttamente proporzionale al rapporto NAD + / NADH mitocondriale, e quindi l'attività ossidoreduttasi BDH1969 modula il potenziale redox mitocondriale (Krebs et al., 1967; Williamson et al., 1929). AcAc può anche decarbossilare spontaneamente in acetone (Pedersen, 7), la fonte di odore dolce negli esseri umani che soffrono di chetoacidosi (cioè, corpi chetonici sierici totali> ~ 3.6 mM; AcAc pKa 4.7,? OHB pKa 1). I meccanismi attraverso i quali i corpi chetonici vengono trasportati attraverso la membrana interna mitocondriale non sono noti, ma AcAc / d-? OHB vengono rilasciati dalle cellule tramite trasportatori monocarbossilati (nei mammiferi, MCT 2 e 16, noti anche come vettori di soluti 1A membri della famiglia 7 e 2011) e trasportati in circolo ai tessuti extraepatici per l'ossidazione terminale (Cotter et al., 2012; Halestrap e Wilson, 2012; Halestrap, 2012; Hugo et al., 1940). Le concentrazioni dei corpi chetonici circolanti sono più alte di quelle nei tessuti extraepatici (Harrison e Long, 1) indicando che i corpi chetonici sono trasportati lungo un gradiente di concentrazione. Le mutazioni con perdita di funzione in MCTXNUMX sono associate ad attacchi spontanei di chetoacidosi, suggerendo un ruolo critico nell'importazione del corpo chetonico.
Ad eccezione della potenziale diversione dei corpi chetonici in destini non ossidativi (vedere Destini metabolici non ossidativi dei corpi chetonici), gli epatociti non hanno la capacità di metabolizzare i corpi chetonici che producono. I corpi chetonici sintetizzati de novo dal fegato sono (i) catabolizzati nei mitocondri dei tessuti extraepatici in acetil-CoA, che è disponibile per il ciclo TCA per l'ossidazione terminale (Fig. 1A), (ii) deviato alle vie di lipogenesi o sintesi degli steroli ( Fig. 1B) o (iii) escreto nelle urine. Come combustibile energetico alternativo, i corpi chetonici sono ossidati avidamente nel cuore, nei muscoli scheletrici e nel cervello (Balasse e Fery, 1989; Bentourkia et al., 2009; Owen et al., 1967; Reichard et al., 1974; Sultan, 1988; ). Il BDH1 mitocondriale extraepatico catalizza la prima reazione di ossidazione? OHB, convertendola in AcAc di ritorno (LEHNINGER et al., 1960; Sandermann et al., 1986). Una d-? OHB-deidrogenasi citoplasmatica (BDH2) con solo il 20% di identità di sequenza per BDH1 ha un elevato Km per i corpi chetonici e svolge anche un ruolo nell'omeostasi del ferro (Davuluri et al., 2016; Guo et al., 2006) . Nella matrice mitocondriale extraepatica, AcAc viene attivato in AcAc-CoA attraverso lo scambio di una frazione CoA da succinil-CoA in una reazione catalizzata da un'unica transferasi CoA di mammifero, succinil-CoA: 3-ossoacido-CoA transferasi (SCOT, CoA transferasi; codificato da OXCT1), attraverso una reazione di quasi equilibrio. L'energia libera rilasciata dall'idrolisi di AcAc-CoA è maggiore di quella del succinil-CoA, favorendo la formazione di AcAc. Pertanto, il flusso ossidativo del corpo chetonico si verifica a causa dell'azione di massa: un abbondante apporto di AcAc e il rapido consumo di acetil-CoA attraverso la citrato sintasi favoriscono la formazione di AcAc-CoA (+ succinato) da parte di SCOT. In particolare, a differenza del glucosio (esochinasi) e degli acidi grassi (sintetasi acil-CoA), l'attivazione dei corpi chetonici (SCOT) in una forma ossidabile non richiede l'investimento di ATP. Una reazione reversibile della tiolasi AcAc-CoA [catalizzata da una qualsiasi delle quattro tiolasi mitocondriali codificate da ACAA2 (che codifica un enzima noto come T1 o CT), ACAT1 (codifica T2), HADHA o HADHB] produce due molecole di acetil-CoA, che entrano nel ciclo TCA (Hersh e Jencks, 1967; Stern et al., 1956; Williamson et al., 1971). Durante gli stati chetotici (cioè, chetoni sierici totali> 500 M), i corpi chetonici contribuiscono in modo significativo al dispendio energetico e vengono utilizzati rapidamente nei tessuti fino a quando si verifica l'assorbimento o la saturazione dell'ossidazione (Balasse et al., 1978; Balasse e Fery, 1989 ; Edmond et al., 1987). Una frazione molto piccola dei corpi chetonici derivati dal fegato può essere prontamente misurata nelle urine e le velocità di utilizzo e riassorbimento da parte del rene sono proporzionate alla concentrazione circolante (Goldstein, 1987; Robinson e Williamson, 1980). Durante gli stati altamente chetotici (> 1 mM nel plasma), la chetonuria funge da reporter semiquantitativo della chetosi, sebbene la maggior parte dei test clinici sui corpi chetonici delle urine rilevi AcAc ma non? OHB (Klocker et al., 2013).
Substrati chetogenici e loro impatto sul metabolismo degli epatociti
I substrati chetogenici comprendono acidi grassi e amminoacidi (Fig. 1B). Il catabolismo degli amminoacidi, in particolare la leucina, genera circa il 4% dei corpi chetonici nello stato postassorbente (Thomas et al., 1982). Pertanto il pool di substrati dell'acetile-CoA per generare corpi chetonici deriva principalmente da acidi grassi, poiché durante gli stati di ridotta assunzione di carboidrati, il piruvato entra nel ciclo epatico del TCA principalmente tramite anaplerosi, cioè carbossilazione ATP-dipendente a ossalacetato (OAA) o malato (MAL) e non decarbossilazione ossidativa in acetil-CoA (Jeoung et al., 2012; Magnusson et al., 1991; Merritt et al., 2011). Nel fegato, il glucosio e il piruvato contribuiscono in modo trascurabile alla chetogenesi, anche quando la piruvato decarbossilazione in acetil-CoA è massima (Jeoung et al., 2012).
L'acetil-CoA ricopre diversi ruoli integrali del metabolismo intermedio epatico oltre la generazione di ATP attraverso l'ossidazione terminale (vedi anche Integrazione del metabolismo del corpo chetone, modificazione post-traduzionale e fisiologia cellulare). L'acetil-CoA attiva allostericamente (i) piruvato carbossilasi (PC), attivando così un meccanismo di controllo metabolico che aumenta l'ingresso anaplerotico dei metaboliti nel ciclo TCA (Owen et al., 2002; Scrutton e Utter, 1967) e (ii) piruvato deidrogenasi chinasi, che fosforila e inibisce la piruvato deidrogenasi (PDH) (Cooper et al., 1975), migliorando così ulteriormente il flusso di piruvato nel ciclo TCA attraverso l'anaplerosi. Inoltre, l'acetil-CoA citoplasmatico, il cui pool è potenziato da meccanismi che convertono l'acetil-CoA mitocondriale in metaboliti trasportabili, inibisce l'ossidazione degli acidi grassi: acetil-CoA carbossilasi (ACC) catalizza la conversione di acetil-CoA in malonil-CoA, il substrato lipogenico e inibitore allosterico del CPT1 mitocondriale [rivisto in (Kahn et al., 2005; McGarry and Foster, 1980)]. Pertanto, il pool di acetil-CoA mitocondriale regola sia ed è regolato dalla via di spillover della chetogenesi, che orchestra aspetti chiave del metabolismo intermedio epatico.
Fatti metabolici non ossidativi dei corpi chetonici
Il destino predominante dei chetoni derivati dal fegato è l'ossidazione extraepatica dipendente dallo SCOT. Tuttavia, AcAc può essere esportato dai mitocondri e utilizzato in percorsi anabolici attraverso la conversione in AcAc-CoA mediante una reazione dipendente dall'ATP catalizzata dalla citetilmetano acetoacetil-CoA sintetasi (AACS, Fig. 1B). Questo percorso è attivo durante lo sviluppo del cervello e nella ghiandola mammaria che allatta (Morris, 2005, Robinson and Williamson, 1978; Ohgami et al., 2003). L'AACS è anche altamente espresso nel tessuto adiposo e negli osteoclasti attivati (Aguilo et al., 2010; Yamasaki et al., 2016). AcAc-CoA citoplasmatico può essere diretto da citosolico HMGCS1 verso la biosintesi dello sterolo, o scisso da due di due tiolasi citoplasmatiche ad acetil-CoA (ACAA1 e ACAT2), carbossilato a malonil-CoA e contribuire alla sintesi di acidi grassi (Bergstrom et al., 1984; Edmond, 1974; Endemann et al., 1982; Geelen et al., 1983; Webber e Edmond, 1977).
Mentre il significato fisiologico deve ancora essere stabilito, i chetoni possono fungere da substrati anabolizzanti anche nel fegato. In contesti sperimentali artificiali, AcAc può contribuire fino alla metà dei lipidi di nuova sintesi e fino al 75% del nuovo colesterolo sintetizzato (Endemann et al., 1982; Geelen et al., 1983; Freed et al., 1988). Poiché AcAc deriva dall'ossidazione incompleta dei grassi epatici, la capacità di AcAc di contribuire alla lipogenesi in vivo implicherebbe un ciclo futile epatico, dove i chetoni derivati dal grasso possono essere utilizzati per la produzione di lipidi, una nozione il cui significato fisiologico richiede una convalida sperimentale, ma potrebbe servire ruoli adattivi o disadattivi (Solinas et al., 2015). AcAc fornisce avidamente la colesterogenesi, con un basso AACS Km-AcAc (~ 50 M) che favorisce l'attivazione di AcAc anche a stomaco pieno (Bergstrom et al., 1984). Il ruolo dinamico del metabolismo dei chetoni citoplasmatici è stato suggerito nei neuroni embrionali primari di topo e negli adipociti derivati 3T3-L1, poiché il knockdown dell'AACS ha alterato la differenziazione di ciascun tipo di cellula (Hasegawa et al., 2012a; Hasegawa et al., 2012b). Il knockdown dell'AACS nei topi in vivo ha ridotto il colesterolo sierico (Hasegawa et al., 2012c). SREBP-2, un regolatore trascrizionale principale della biosintesi del colesterolo e recettore attivato dal proliferatore del perossisoma (PPAR) -? sono attivatori trascrizionali dell'AACS e regolano la sua trascrizione durante lo sviluppo dei neuriti e nel fegato (Aguilo et al., 2010; Hasegawa et al., 2012c). Nel loro insieme, il metabolismo del corpo chetonico citoplasmatico può essere importante in determinate condizioni o storie naturali di malattie, ma è inadeguato per smaltire i corpi chetonici derivati dal fegato, poiché si verifica una massiccia iperchetonemia nel contesto di una compromissione selettiva del destino ossidativo primario attraverso la perdita di mutazioni funzionali a SCOT (Berry et al., 2001; Cotter et al., 2011).
Regolazione di HMGCS2 e SCOT / OXCT1
La divergenza di un mitocondrio dal gene che codifica HMGCS citosolico si è manifestata precocemente nell'evoluzione dei vertebrati a causa della necessità di supportare la chetogenesi epatica in specie con rapporti tra il cervello e il peso corporeo più elevati (Boukaftane et al., 1994; Cunnane and Crawford, 2003). Le mutazioni HMGCS2 di perdita naturale di funzione nell'uomo causano attacchi di ipoglicemia ipoketotica (Pitt et al., 2015; Thompson et al., 1997). L'espressione robusta HMGCS2 è limitata agli epatociti e all'epitelio del colon, e la sua espressione e attività enzimatica sono coordinate attraverso diversi meccanismi (Mascaro et al., 1995; McGarry e Foster, 1980; Robinson e Williamson, 1980). Mentre l'intera gamma di stati fisiologici che influenzano HMGCS2 richiede un'ulteriore delucidazione, la sua espressione e / o attività è regolata durante il periodo postnatale, invecchiamento, diabete, fame o ingestione di dieta chetogenica (Balasse e Fery, 1989; Cahill GF Jr, 2006 ; Girard et al., 1992; Hegardt, 1999; Satapati et al., 2012; Sengupta et al., 2010). Nel feto, la metilazione della regione fiancheggiante 5 del gene Hmgcs2 è inversamente correlata alla sua trascrizione ed è parzialmente invertita dopo la nascita (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al. ., 1983). Allo stesso modo, il Bdh1 epatico mostra un pattern di espressione evolutivo, crescente dalla nascita allo svezzamento, ed è anche indotto dalla dieta chetogenica in un fattore di crescita dei fibroblasti (FGF) -21-dipendente (Badman et al., 2007; Zhang et al., 1989 ). La chetogenesi nei mammiferi è altamente reattiva sia per l'insulina che per il glucagone, essendo soppressa e stimolata, rispettivamente (McGarry e Foster, 1977). L'insulina sopprime la lipolisi del tessuto adiposo, privando così la chetogenesi del suo substrato, mentre il glucagone aumenta il flusso chetogenico attraverso un effetto diretto sul fegato (Hegardt, 1999). La trascrizione di Hmgcs2 è stimolata dal fattore di trascrizione forkhead FOXA2, che viene inibito tramite insulina-fosfatidilinositolo-3-chinasi / Akt ed è indotto dal segnale glucagone-cAMP-p300 (Arias et al., 1995; Hegardt, 1999; Quant et al. , 1990; Thumelin et al., 1993; von Meyenn et al., 2013; Wolfrum et al., 2004; Wolfrum et al., 2003). PPAR? (Rodriguez et al., 1994) insieme al suo target, FGF21 (Badman et al., 2007) inducono anche la trascrizione di Hmgcs2 nel fegato durante la fame o la somministrazione di dieta chetogenica (Badman et al., 2007; Inagaki et al., 2007 ). Induzione di PPAR? può verificarsi prima del passaggio dalla fisiologia fetale a quella neonatale, mentre l'attivazione di FGF21 può essere favorita nel periodo neonatale precoce tramite l'inibizione mediata da? OHB dell'istone deacetilasi (HDAC) -3 (Rando et al., 2016). Inibizione dipendente da mTORC1 (mammifero bersaglio del complesso rapamicina 1) di PPAR? l'attività trascrizionale è anche un regolatore chiave dell'espressione genica di Hmgcs2 (Sengupta et al., 2010) e il PER2 epatico, un oscillatore circadiano principale, regola indirettamente l'espressione di Hmgcs2 (Chavan et al., 2016). Recenti osservazioni indicano che l'interleuchina-6 extraepatica indotta dal tumore altera la chetogenesi tramite PPAR? soppressione (Flint et al., 2016).
L'attività dell'enzima HMGCS2 è regolata tramite più PTM. La fosforilazione di serina HMGCS2 ha potenziato la sua attività in vitro (Grimsrud et al., 2012). L'attività di HMGCS2 è inibita allostericamente dalla succinilazione di succinil-CoA e residui di lisina (Arias et al., 1995; Hegardt, 1999; Lowe and Tubbs, 1985; Quant et al., 1990; Rardin et al., 2013; Reed et al., 1975; Thumelin et al., 1993). La succinilazione dei residui di lisina HMGCS2, HMGCL e BDH1 nei mitocondri epatici sono bersagli del deacilasi sirtuin dipendente da NAD + 5 (SIRT5) (Rardin et al., 2013). L'attività di HMGCS2 è anche potenziata dalla deacetilazione della lisina SIRT3 ed è possibile che la diafonia tra acetilazione e succinilazione regoli l'attività di HMGCS2 (Rardin et al., 2013; Shimazu et al., 2013). Nonostante la capacità di questi PTM di regolare HMGCS2 Km e Vmax, le fluttuazioni di questi PTM non sono state ancora accuratamente mappate e non sono state confermate come driver meccanicistici della chetogenesi in vivo.
SCOT è espresso in tutte le cellule di mammiferi che ospitano i mitocondri, ad eccezione di quelli degli epatociti. L'importanza dell'attività di SCOT e della chetolisi è stata dimostrata nei topi SCOT-KO, che hanno presentato una letalità uniforme a causa dell'ipoglicemia iperchetonemia entro 48h dopo la nascita (Cotter et al., 2011). La perdita di SCOT dei tessuti nei neuroni o nei miociti scheletrici induce anomalie metaboliche durante la fame ma non è letale (Cotter et al., 2013b). Nell'uomo il deficit di SCOT si manifesta precocemente nella vita con chetoacidosi grave, che causa letargia, vomito e coma (Berry et al., 2001; Fukao et al., 2000; Kassovska-Bratinova et al., 1996; Niezen-Koning et al. , 1997; Saudubray et al., 1987; Snyderman et al., 1998; Tildon e Cornblath, 1972). Relativamente poco è noto a livello cellulare sui regolatori di espressione di geni e proteine SCOT. L'espressione dell'mRNA di Oxct1 e la proteina e l'attività di SCOT sono diminuite negli stati chetotici, possibilmente attraverso meccanismi PPAR-dipendenti (Fenselau e Wallis, 1974; Fenselau e Wallis, 1976; Grinblat et al., 1986; Okuda et al., 1991; Turko et al ., 2001; Wentz et al., 2010). Nella chetoacidosi diabetica, la mancata corrispondenza tra chetogenesi epatica ed ossidazione extraepatica si acuisce a causa della compromissione dell'attività di SCOT. La sovraespressione del trasportatore di glucosio insulino-indipendente (GLUT1 / SLC2A1) nei cardiomiociti inibisce anche l'espressione del gene Oxct1 e downregula l'ossidazione terminale dei chetoni in uno stato non chetotico (Yan et al., 2009). Nel fegato, l'abbondanza di mRNA di Oxct1 è soppressa da microRNA-122 e metilazione dell'istone H3K27me3 che sono evidenti durante la transizione dal periodo fetale a quello neonatale (Thorrez et al., 2011). Tuttavia, la soppressione dell'espressione di Oxct1 epatica nel periodo postnatale è principalmente attribuibile all'evacuazione di progenitori ematopoietici che esprimono Oxct1 dal fegato, piuttosto che alla perdita dell'espressione di Oxct1 preesistente in epatociti differenziati terminalmente. Infatti, l'espressione dell'mRNA di Oxct1 e della proteina SCOT negli epatociti differenziati è estremamente bassa (Orii et al., 2008).
SCOT è anche regolamentato da PTM. L'enzima è iperacetilato nel cervello dei topi SIRT3 KO, che mostrano anche una ridotta produzione di acetil-CoA dipendente da AcAc (Dittenhafer-Reed et al., 2015). La nitrazione non enzimatica dei residui di tirosina di SCOT ne attenua anche l'attività, che è stata segnalata nei cuori di vari modelli di topi diabetici (Marcondes et al., 2001; Turko et al., 2001; Wang et al., 2010a). Al contrario, la nitrazione dei residui di triptofano aumenta l'attività degli SCOT (Brégére et al., 2010; Rebrin et al., 2007). Possono esistere meccanismi molecolari di nitrazione o de-nitrazione specifici del residuo progettati per modulare l'attività SCOT e richiedono delucidazione.
Controversie nella chetogenesi extraepatica
Nei mammiferi l'organo chetogenico primario è il fegato e solo gli epatociti e le cellule epiteliali intestinali esprimono abbondantemente l'isoforma mitocondriale di HMGCS2 (Cotter et al., 2013a; Cotter et al., 2014; McGarry e Foster, 1980; Robinson e Williamson, 1980) . La fermentazione batterica anaerobica di polisaccaridi complessi produce butirrato, che viene assorbito dai colonociti nei mammiferi per ossidazione terminale o chetogenesi (Cherbuy et al., 1995), che può svolgere un ruolo nella differenziazione dei colonociti (Wang et al., 2016). Escludendo le cellule epiteliali intestinali e gli epatociti, l'HMGCS2 è quasi assente in quasi tutte le altre cellule di mammifero, ma la prospettiva di chetogenesi extraepatica è stata sollevata nelle cellule tumorali, negli astrociti del sistema nervoso centrale, nel rene, nel pancreas? cellule, epitelio pigmentato retinico (RPE) e persino nel muscolo scheletrico (Adijanto et al., 2014; Avogaro et al., 1992; El Azzouny et al., 2016; Grabacka et al., 2016; Kang et al., 2015 ; Le Foll et al., 2014; Nonaka et al., 2016; Takagi et al., 2016a; Thevenet et al., 2016; Zhang et al., 2011). HMGCS2 ectopico è stato osservato in tessuti privi di capacità chetogenica netta (Cook et al., 2016; Wentz et al., 2010) e HMGCS2 mostra attività prospettiche di `` moonlighting '' indipendenti dalla chetogenesi, anche all'interno del nucleo cellulare (Chen et al. , 2016; Kostiuk et al., 2010; Meertens et al., 1998).
Qualsiasi tessuto extraepatico che ossida i corpi chetonici ha anche il potenziale per accumulare corpi chetonici tramite meccanismi indipendenti HMGCS2 (Fig. 2A). Tuttavia, non esiste tessuto extraepatico in cui una concentrazione corporea di chetoni allo stato stazionario superi quella nella circolazione (Cotter et al., 2011; Cotter et al., 2013b; Harrison e Long, 1940), sottolineando che i corpi chetonici vengono trasportati gradiente di concentrazione tramite meccanismi MCT1 / 2-dipendenti. Un meccanismo di apparente chetogenesi extraepatica può effettivamente riflettere una relativa compromissione dell'ossidazione chetonica. Ulteriori potenziali spiegazioni rientrano nel regno della formazione del corpo chetonico. In primo luogo, la chetogenesi de novo può verificarsi tramite l'attività enzimatica reversibile della tiolasi e della SCOT (Weidemann e Krebs, 1969). Quando la concentrazione di acetil-CoA è relativamente alta, le reazioni normalmente responsabili dell'ossidazione di AcAc operano nella direzione opposta (GOLDMAN, 1954). Un secondo meccanismo si verifica quando gli intermedi derivati dall'ossidazione si accumulano a causa di un collo di bottiglia del ciclo TCA, AcAc-CoA viene convertito in l-? OHB-CoA attraverso una reazione catalizzata dalla 3-idrossiacil-CoA deidrogenasi mitocondriale e ulteriormente da 3-idrossibutirile CoA deacylase a l-? OHB, che è indistinguibile mediante spettrometria di massa o spettroscopia di risonanza dall'enantiomero fisiologico d-? OHB (Reed e Ozand, 1980). l-? OHB può essere distinto cromatograficamente o enzimaticamente da d-? OHB ed è presente nei tessuti extraepatici, ma non nel fegato o nel sangue (Hsu et al., 2011). La chetogenesi epatica produce solo d-? OHB, l'unico enantiomero che è un substrato BDH (Ito et al., 1984; Lincoln et al., 1987; Reed and Ozand, 1980; Scofield et al., 1982; Scofield et al., 1982). Un terzo meccanismo indipendente da HMGCS2 genera d-? OHB attraverso il catabolismo degli amminoacidi, in particolare quello della leucina e della lisina. Un quarto meccanismo è apparente solo perché è dovuto a un artefatto di etichettatura ed è quindi definito pseudoketogenesis. Questo fenomeno è attribuibile alla reversibilità delle reazioni SCOT e tiolasi e può causare una sovrastima del turnover corporeo chetonico a causa della diluizione isotopica del tracciante corporeo chetonico nel tessuto extraepatico (Des Rosiers et al., 1990; Fink et al., 1988) . Tuttavia, la pseudochetogenesi può essere trascurabile nella maggior parte dei contesti (Bailey et al., 1990; Keller et al., 1978). Uno schema (Fig. 2A) indica un approccio utile da applicare considerando un'elevata concentrazione tissutale di chetoni allo stato stazionario.
Il rene è stato recentemente oggetto di attenzione come organo potenzialmente chetogenico. Nella stragrande maggioranza degli stati, il rene è un consumatore netto di corpi chetonici derivati dal fegato, che espellono o riassorbono corpi chetonici dal flusso sanguigno, e il rene generalmente non è un generatore o concentratore di corpi chetonici netti (Robinson e Williamson, 1980). Gli autori di uno studio classico hanno concluso che la chetogenesi renale minima quantificata in un sistema sperimentale artificiale non era fisiologicamente rilevante (Weidemann e Krebs, 1969). Recentemente, la chetogenesi renale è stata dedotta in modelli murini diabetici e con deficit di autofagia, ma è più probabile che cambiamenti multiorgano nell'omeostasi metabolica alterino il metabolismo chetonico integrativo attraverso input su più organi (Takagi et al., 2016a; Takagi et al., 2016b; Zhang et al., 2011). Una recente pubblicazione ha suggerito la chetogenesi renale come meccanismo protettivo contro il danno da ischemia-riperfusione nel rene (Tran et al., 2016). Le concentrazioni assolute allo stato stazionario di? OHB da estratti di tessuto renale di topo sono state riportate a ~ 4-12 mM. Per verificare se questo era sostenibile, abbiamo quantificato le concentrazioni di? OHB in estratti renali da topi alimentati e 24 ore a digiuno. Le concentrazioni di? OHB nel siero sono aumentate da ~ 100 M a 2 mM con 24 ore di digiuno (Fig. 2B), mentre le concentrazioni renali di? OHB allo stato stazionario si avvicinano a 100 M a stomaco pieno e solo 1 mM a 24 ore di digiuno (Fig. 2C E), osservazioni coerenti con le concentrazioni quantificate oltre 45 anni fa (Hems e Brosnan, 1970). Resta possibile che negli stati chetotici, i corpi chetonici derivati dal fegato possano essere renoprotettivi, ma l'evidenza della chetogenesi renale richiede ulteriori prove. Prove convincenti che supportano la vera chetogenesi extraepatica sono state presentate in RPE (Adijanto et al., 2014). Questa intrigante trasformazione metabolica è stata suggerita per consentire potenzialmente ai chetoni derivati dall'RPE di fluire al fotorecettore o alle cellule della glia di Mller, che potrebbero aiutare nella rigenerazione del segmento esterno del fotorecettore.
? OHB come mediatore di segnalazione
Sebbene siano energeticamente ricchi, i corpi chetonici esercitano ruoli di segnalazione provocatori `` non canonici '' nell'omeostasi cellulare (Fig.3) (Newman e Verdin, 2014; Rojas-Morales et al., 2016). Ad esempio,? OHB inibisce gli HDAC di Classe I, che aumenta l'acetilazione degli istoni e quindi induce l'espressione di geni che riducono lo stress ossidativo (Shimazu et al., 2013). ? L'OHB stesso è un modificatore covalente dell'istone dei residui di lisina nel fegato di topi diabetici a digiuno o indotti da streptozotocina (Xie et al., 2016) (vedere anche di seguito, Integrazione del metabolismo corporeo chetonico, modificazione post-traduzionale e fisiologia cellulare, e Corpi chetonici, stress ossidativo e neuroprotezione).
ï ¿½
? L'OHB è anche un effettore tramite i recettori accoppiati alla proteina G. Attraverso meccanismi molecolari poco chiari, sopprime l'attività del sistema nervoso simpatico e riduce il dispendio energetico totale e la frequenza cardiaca inibendo la segnalazione degli acidi grassi a catena corta attraverso il recettore accoppiato alla proteina G 41 (GPR41) (Kimura et al., 2011). Uno degli effetti di segnalazione più studiati di? OHB procede attraverso GPR109A (noto anche come HCAR2), un membro della sottofamiglia GPCR dell'acido idrocarbossilico espresso nei tessuti adiposi (bianchi e marroni) (Tunaru et al., 2003), e in cellule immunitarie (Ahmed et al., 2009). ? OHB è l'unico ligando endogeno noto del recettore GPR109A (EC50 ~ 770 M) attivato da d-? OHB, l-? OHB e butirrato, ma non AcAc (Taggart et al., 2005). L'elevata soglia di concentrazione per l'attivazione di GPR109A viene raggiunta attraverso l'adesione a una dieta chetogenica, fame o durante la chetoacidosi, portando all'inibizione della lipolisi del tessuto adiposo. L'effetto anti-lipolitico di GPR109A procede attraverso l'inibizione dell'adenil ciclasi e della diminuzione del cAMP, inibendo la lipasi trigliceridica sensibile agli ormoni (Ahmed et al., 2009; Tunaru et al., 2003). Questo crea un ciclo di feedback negativo in cui la chetosi pone un freno modulatorio alla chetogenesi diminuendo il rilascio di acidi grassi non esterificati dagli adipociti (Ahmed et al., 2009; Taggart et al., 2005), un effetto che può essere controbilanciato da la spinta simpatica che stimola la lipolisi. La niacina (vitamina B3, acido nicotinico) è un potente ligando (EC50 ~ 0.1 M) per GRP109A, efficacemente impiegato per decenni per le dislipidemie (Benyo et al., 2005; Benyo et al., 2006; Fabbrini et al., 2010a; Lukasova et al., 2011; Tunaru et al., 2003). Mentre la niacina aumenta il trasporto inverso del colesterolo nei macrofagi e riduce le lesioni aterosclerotiche (Lukasova et al., 2011), gli effetti di? OHB sulle lesioni aterosclerotiche rimangono sconosciuti. Sebbene il recettore GPR109A eserciti ruoli protettivi ed esistano connessioni intriganti tra l'uso della dieta chetogenica nell'ictus e nelle malattie neurodegenerative (Fu et al., 2015; Rahman et al., 2014), un ruolo protettivo di? OHB tramite GPR109A non è stato dimostrato in vivo .
Infine,? OHB può influenzare l'appetito e la sazietà. Una meta-analisi di studi che hanno misurato gli effetti delle diete chetogeniche ea bassissimo contenuto energetico ha concluso che i partecipanti che consumano queste diete mostrano una maggiore sazietà, rispetto alle diete di controllo (Gibson et al., 2015). Tuttavia, una spiegazione plausibile per questo effetto sono gli elementi metabolici o ormonali aggiuntivi che potrebbero modulare l'appetito. Ad esempio, i topi mantenuti con una dieta chetogenica di roditori hanno mostrato un aumento del dispendio energetico rispetto ai topi nutriti con chow control, nonostante un apporto calorico simile, e la leptina circolante oi geni dei peptidi che regolano il comportamento alimentare non sono stati modificati (Kennedy et al., 2007). Tra i meccanismi proposti che suggeriscono la soppressione dell'appetito da? OHB include sia la segnalazione che l'ossidazione (Laeger et al., 2010). La delezione specifica degli epatociti del gene del ritmo circadiano (Per2) e studi di immunoprecipitazione della cromatina hanno rivelato che PER2 attiva direttamente il gene Cpt1a e regola indirettamente Hmgcs2, portando a chetosi alterata nei topi knockout per Per2 (Chavan et al., 2016). Questi topi hanno mostrato una ridotta anticipazione del cibo, che è stata parzialmente ripristinata dalla somministrazione sistemica di? OHB. Saranno necessari studi futuri per confermare il sistema nervoso centrale come bersaglio diretto? OHB e se l'ossidazione chetonica è necessaria per gli effetti osservati, o se è coinvolto un altro meccanismo di segnalazione. Altri ricercatori hanno invocato la possibilità di una chetogenesi derivata dagli astrociti locale all'interno dell'ipotalamo ventromediale come regolatore dell'assunzione di cibo, ma queste osservazioni preliminari beneficeranno anche di valutazioni genetiche e basate sul flusso (Le Foll et al., 2014). La relazione tra chetosi e mancanza di nutrienti rimane interessante perché la fame e la sazietà sono elementi importanti nei tentativi di perdita di peso falliti.
Integrazione del metabolismo corporeo chetonico, modificazione post-traduzionale e fisiologia cellulare
I corpi chetonici contribuiscono a compartimenti stagni di acetil-CoA, un intermedio chiave che presenta ruoli importanti nel metabolismo cellulare (Pietrocola et al., 2015). Un ruolo dell'acetil-CoA è quello di fungere da substrato per l'acetilazione, una modifica covalente istonica enzimaticamente catalizzata (Choudhary et al., 2014; Dutta et al., 2016; Fan et al., 2015; Menzies et al., 2016 ). Un gran numero di proteine mitocondriali dinamicamente acetilate, molte delle quali possono verificarsi attraverso meccanismi non enzimatici, sono emerse anche da studi di proteomica computazionale (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013 Shimazu et al., 2010). Le deacetilasi di lisina usano un cofattore di zinco (ad esempio, HDAC nucleocitosolici) o NAD + come co-substrato (sirtuine, SIRT) (Choudhary et al., 2014; Menzies et al., 2016). L'acetilproteoma funge sia da sensore che da effettore del pool di acetil-CoA cellulare totale, poiché le manipolazioni fisiologiche e genetiche portano ciascuna a variazioni globali non enzimatiche di acetilazione (Weinert et al., 2014). Poiché i metaboliti intracellulari fungono da modulatori dell'acetilazione dei residui di lisina, è importante considerare il ruolo dei corpi chetonici, la cui abbondanza è altamente dinamica.
? OHB è un modificatore epigenetico attraverso almeno due meccanismi. Livelli aumentati di? OHB indotti da digiuno, restrizione calorica, somministrazione diretta o esercizio prolungato provocano l'inibizione di HDAC o l'attivazione dell'istone acetiltransferasi (Marosi et al., 2016; Sleiman et al., 2016) o allo stress ossidativo (Shimazu et al., 2013) . ? L'inibizione dell'OHB di HDAC3 potrebbe regolare la fisiologia metabolica del neonato (Rando et al., 2016). Indipendentemente, lo stesso? OHB modifica direttamente i residui di lisina istonica (Xie et al., 2016). Il digiuno prolungato o la chetoacidosi diabetica indotta dalla steptozotocina hanno aumentato l'istone? -Idrossibutirilazione. Sebbene il numero di siti di lisina-idrossibutirilazione e acetilazione fosse paragonabile, è stata osservata stechiometricamente maggiore istone-idrossibutirilazione rispetto all'acetilazione. Geni distinti sono stati influenzati dall'istone lisina? -Idrossibutirilazione, rispetto all'acetilazione o alla metilazione, suggerendo funzioni cellulari distinte. Non è noto se la? -Idrossibutirilazione sia spontanea o enzimatica, ma espande la gamma di meccanismi attraverso i corpi chetonici che influenzano dinamicamente la trascrizione.
Gli eventi di riprogrammazione cellulare essenziale durante la restrizione calorica e la privazione dei nutrienti possono essere mediati nella deacetilazione mitocondriale e nella desuccinilazione dipendenti da SIRT3 e SIRT5, rispettivamente, regolando le proteine chetogeniche e chetolitiche a livello post-traduzionale nel fegato e nei tessuti extraepatici (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013; Shimazu et al., 2010). Anche se il confronto stechiometrico dei siti occupati non si collega necessariamente direttamente ai cambiamenti nel flusso metabolico, l'acetilazione mitocondriale è dinamica e può essere guidata dalla concentrazione di acetil-CoA o dal pH mitocondriale, piuttosto che dalle acetiltransferasi enzimatiche (Wagner e Payne, 2013). Il fatto che SIRT3 e SIRT5 modulino le attività degli enzimi che metabolizzano il corpo chetonico provoca la questione del ruolo reciproco dei chetoni nello scolpire l'acetilproteoma, il succinilproteoma e altri bersagli cellulari dinamici. Infatti, poiché le variazioni della chetogenesi riflettono le concentrazioni di NAD +, la produzione e l'abbondanza di chetoni potrebbero regolare l'attività della sirtuina, influenzando così i pool totali di acetil-CoA / succinil-CoA, l'acilproteoma e quindi la fisiologia mitocondriale e cellulare. ? -idrossibutirilazione dei residui di lisina enzimatica potrebbe aggiungere un altro strato alla riprogrammazione cellulare. Nei tessuti extraepatici, l'ossidazione del corpo chetonico può stimolare analoghi cambiamenti nell'omeostasi cellulare. Mentre la compartimentazione dei pool di acetil-CoA è altamente regolata e coordina un ampio spettro di cambiamenti cellulari, la capacità dei corpi chetonici di modellare direttamente le concentrazioni di acetil-CoA mitocondriali e citoplasmatiche richiede delucidazione (Chen et al., 2012; Corbet et al., 2016; Pougovkina et al., 2014; Schwer et al., 2009; Wellen e Thompson, 2012). Poiché le concentrazioni di acetil-CoA sono strettamente regolate e l'acetil-CoA è impermeabile alla membrana, è fondamentale considerare i meccanismi driver che coordinano l'omeostasi dell'acetil-CoA, inclusi i tassi di produzione e l'ossidazione terminale nel ciclo TCA, la conversione in corpi chetonici, mitocondriale efflusso tramite carnitina acetiltransferasi (CrAT) o esportazione di acetil-CoA in citosol dopo la conversione in citrato e il rilascio da parte dell'ATP citrato liasi (ACLY). I ruoli chiave di questi ultimi meccanismi nell'acetilproteoma cellulare e nell'omeostasi richiedono una comprensione corrispondente dei ruoli della chetogenesi e dell'ossidazione chetonica (Das et al., 2015; McDonnell et al., 2016; Moussaieff et al., 2015; Overmyer et al., 2015; Seiler et al., 2014; Seiler et al., 2015; Wellen et al., 2009; Wellen e Thompson, 2012). Saranno necessarie tecnologie convergenti in metabolomica e acilproteomica nell'impostazione di modelli geneticamente manipolati per specificare obiettivi e risultati.
Risposte anti- e pro-infiammatorie ai corpi chetonici
La chetosi e i corpi chetonici modulano l'infiammazione e la funzione delle cellule immunitarie, ma sono stati proposti meccanismi vari e persino discrepanti. La privazione prolungata di nutrienti riduce l'infiammazione (Youm et al., 2015), ma la chetosi cronica del diabete di tipo 1 è uno stato pro-infiammatorio (Jain et al., 2002; Kanikarla-Marie e Jain, 2015; Kurepa et al., 2012 ). I ruoli di segnalazione basati sul meccanismo per? OHB nell'infiammazione emergono perché molte cellule del sistema immunitario, inclusi macrofagi o monociti, esprimono abbondantemente GPR109A. Mentre? OHB esercita una risposta prevalentemente antinfiammatoria (Fu et al., 2014; Gambhir et al., 2012; Rahman et al., 2014; Youm et al., 2015), alte concentrazioni di corpi chetonici, in particolare AcAc, possono innescare una risposta pro-infiammatoria (Jain et al., 2002; Kanikarla-Marie e Jain, 2015; Kurepa et al., 2012).
I ruoli antinfiammatori dei ligandi GPR109A nell'aterosclerosi, nell'obesità, nelle malattie infiammatorie intestinali, nelle malattie neurologiche e nel cancro sono stati esaminati (Graff et al., 2016). L'espressione di GPR109A è aumentata nelle cellule RPE di modelli diabetici, pazienti diabetici umani (Gambhir et al., 2012) e nella microglia durante la neurodegenerazione (Fu et al., 2014). Gli effetti anti-infiammatori di? OHB sono potenziati dalla sovraespressione di GPR109A nelle cellule RPE e abrogati dall'inibizione farmacologica o dal knockout genetico di GPR109A (Gambhir et al., 2012). ? OHB e acido nicotinico esogeno (Taggart et al., 2005), entrambi conferiscono effetti antinfiammatori nel TNF? o infiammazione indotta da LPS diminuendo i livelli di proteine pro-infiammatorie (iNOS, COX-2) o citochine secrete (TNF ?, IL-1 ?, IL-6, CCL2 / MCP-1), in parte attraverso l'inibizione di NF -? B traslocazione (Fu et al., 2014; Gambhir et al., 2012). ? L'OHB riduce lo stress ER e l'inflammasoma NLRP3, attivando la risposta allo stress antiossidante (Bae et al., 2016; Youm et al., 2015). Tuttavia, nell'infiammazione neurodegenerativa, la protezione mediata da? OHB dipendente da GPR109A non coinvolge mediatori infiammatori come la segnalazione della via MAPK (p. Es., ERK, JNK, p38) (Fu et al., 2014), ma può richiedere PGD1 COX-2-dipendente produzione (Rahman et al., 2014). È interessante che il macrofago GPR109A sia necessario per esercitare un effetto neuroprotettivo in un modello di ictus ischemico (Rahman et al., 2014), ma la capacità di? OHB di inibire l'inflammasoma NLRP3 nei macrofagi derivati dal midollo osseo è GPR109A indipendente (Youm et al. ., 2015). Sebbene la maggior parte degli studi colleghi? OHB agli effetti antinfiammatori,? OHB può essere pro-infiammatorio e aumentare i marcatori di perossidazione lipidica negli epatociti di vitello (Shi et al., 2014). Gli effetti anti-infiammatori di? OHB possono quindi dipendere dal tipo di cellula, dalla concentrazione di? OHB, dalla durata dell'esposizione e dalla presenza o assenza di co-modulatori.
A differenza di? OHB, AcAc può attivare la segnalazione pro-infiammatoria. AcAc elevata, specialmente con un'alta concentrazione di glucosio, intensifica il danno delle cellule endoteliali attraverso un meccanismo dipendente dalla NADPH ossidasi / stress ossidativo (Kanikarla-Marie e Jain, 2015). Elevate concentrazioni di AcAc nel cordone ombelicale di madri diabetiche erano correlate con un più alto tasso di ossidazione delle proteine e concentrazione di MCP-1 (Kurepa et al., 2012). L'alto AcAc nei pazienti diabetici era correlato al TNF? espressione (Jain et al., 2002) e AcAc, ma non? OHB, TNF indotta, espressione di MCP-1, accumulo di ROS e diminuzione del livello di cAMP nelle cellule monocite umane U937 (Jain et al., 2002; Kurepa et al ., 2012).
I fenomeni di segnalazione dipendenti dal corpo chetonico sono spesso innescati solo con concentrazioni corporee di chetoni elevate (> 5 mM) e, nel caso di molti studi che collegano i chetoni a effetti pro o antinfiammatori, attraverso meccanismi poco chiari. Inoltre, a causa degli effetti contraddittori di? OHB contro AcAc sull'infiammazione e della capacità del rapporto AcAc /? OHB di influenzare il potenziale redox mitocondriale, i migliori esperimenti che valutano i ruoli dei corpi chetonici sui fenotipi cellulari confrontano gli effetti di AcAc e? OHB in rapporti variabili e concentrazioni cumulative variabili [ad esempio, (Saito et al., 2016)]. Infine, AcAc può essere acquistato in commercio solo come sale di litio o come estere etilico che richiede l'idrolisi di base prima dell'uso. Il catione di litio induce indipendentemente cascate di trasduzione del segnale (Manji et al., 1995) e l'anione AcAc è labile. Infine, gli studi che utilizzano d / l-? OHB racemico possono essere confusi, poiché solo lo stereoisomero d-? OHB può essere ossidato ad AcAc, ma d-? OHB e l-? OHB possono entrambi segnalare attraverso GPR109A, inibire l'inflammasoma NLRP3, e servono come substrati lipogenici.
Corpi chetonici, stress ossidativo e neuroprotezione
Lo stress ossidativo è tipicamente definito come uno stato in cui i ROS si presentano in eccesso, a causa di un'eccessiva produzione e / o di una ridotta eliminazione. I ruoli antiossidanti e di mitigazione dello stress ossidativo dei corpi chetonici sono stati ampiamente descritti sia in vitro che in vivo, in particolare nel contesto della neuroprotezione. Poiché la maggior parte dei neuroni non genera efficacemente fosfati ad alta energia dagli acidi grassi, ma ossidano i corpi chetonici quando i carboidrati scarseggiano, gli effetti neuroprotettivi dei corpi chetonici sono particolarmente importanti (Cahill GF Jr, 2006; Edmond et al., 1987; Yang et al., 1987). Nei modelli di stress ossidativo, l'induzione BDH1 e la soppressione SCOT suggeriscono che il metabolismo del corpo chetone può essere riprogrammato per sostenere diversi segnali cellulari, potenziale redox o requisiti metabolici (Nagao et al., 2016; Tieu et al., 2003).
I corpi chetonici riducono i gradi di danno cellulare, lesioni, morte e apoptosi inferiore nei neuroni e nei cardiomiociti (Haces et al., 2008; Maalouf et al., 2007; Nagao et al., 2016; Tieu et al., 2003). I meccanismi invocati sono vari e non sempre correlati linearmente alla concentrazione. Basse concentrazioni millimolari di (do l) -? OHB eliminano ROS (anione idrossile), mentre AcAc elimina numerose specie di ROS, ma solo a concentrazioni che superano l'intervallo fisiologico (IC50 20 mM) (Haces et al., 67) . Al contrario, un'influenza benefica sul potenziale redox della catena di trasporto degli elettroni è un meccanismo comunemente collegato al d-? OHB. Mentre tutti e tre i corpi chetonici (d / l-? OHB e AcAc) hanno ridotto la morte delle cellule neuronali e l'accumulo di ROS innescato dall'inibizione chimica della glicolisi, solo d-? OHB e AcAc hanno prevenuto il declino dell'ATP neuronale. Al contrario, in un modello ipoglicemico in vivo, (do l) -? OHB, ma non AcAc ha impedito la perossidazione lipidica ippocampale (Haces et al., 2008; Maalouf et al., 2008; Marosi et al., 2007; Murphy, 2016 ; Tieu et al., 2009). Studi in vivo su topi alimentati con una dieta chetogenica (2003% kcal di grassi e 87% di proteine) hanno mostrato variazioni neuroanatomiche della capacità antiossidante (Ziegler et al., 13), dove i cambiamenti più profondi sono stati osservati nell'ippocampo, con aumento della glutatione perossidasi e totale capacità antiossidanti.
Dieta chetogenica, esteri chetonici (vedi anche Uso terapeutico di dieta chetogenica e corpi chetonici esogeni), o somministrazione di? OHB esercitano neuroprotezione in modelli di ictus ischemico (Rahman et al., 2014); Morbo di Parkinson (Tieu et al., 2003); sequestro di tossicità da ossigeno del sistema nervoso centrale (D'Agostino et al., 2013); spasmi epilettici (Yum et al., 2015); Encefalomiopatia mitocondriale, acidosi lattica e sindrome da episodi di ictus (MELAS) (Frey et al., 2016) e malattia di Alzheimer (Cunnane e Crawford, 2003; Yin et al., 2016). Al contrario, un recente rapporto ha dimostrato prove istopatologiche di progressione neurodegenerativa da una dieta chetogenica in un modello murino transgenico di riparazione anormale del DNA mitocondriale, nonostante l'aumento della biogenesi mitocondriale e delle firme antiossidanti (Lauritzen et al., 2016). Altri rapporti contrastanti suggeriscono che l'esposizione ad alte concentrazioni corporee di chetoni provoca stress ossidativo. Dosi elevate di? OHB o AcAc hanno indotto secrezione di ossido nitrico, perossidazione lipidica, ridotta espressione di SOD, glutatione perossidasi e catalasi negli epatociti di vitello, mentre negli epatociti di ratto l'induzione della via MAPK è stata attribuita ad AcAc ma non? OHB (Abdelmegeed et al., 2004 ; Shi et al., 2014; Shi et al., 2016).
Presi insieme, la maggior parte dei rapporti collega? OHB all'attenuazione dello stress ossidativo, poiché la sua somministrazione inibisce la produzione di ROS / superossido, previene la perossidazione lipidica e l'ossidazione delle proteine, aumenta i livelli di proteine antiossidanti e migliora la respirazione mitocondriale e la produzione di ATP (Abdelmegeed et al., 2004; Haces et al., 2008; Jain et al., 1998; Jain et al., 2002; Kanikarla-Marie e Jain, 2015; Maalouf et al., 2007; Maalouf e Rho, 2008; Marosi et al., 2016; Tieu et al., 2003; Yin et al., 2016; Ziegler et al., 2003). Sebbene l'AcAc sia stato correlato più direttamente di? OHB con l'induzione dello stress ossidativo, questi effetti non sono sempre facilmente sezionabili dalle risposte pro-infiammatorie prospettiche (Jain et al., 2002; Kanikarla-Marie e Jain, 2015; Kanikarla-Marie e Jain, 2016). Inoltre, è fondamentale considerare che l'apparente beneficio antiossidante conferito dalle diete chetogeniche pleiotropiche potrebbe non essere trasdotto dagli stessi corpi chetonici e la neuroprotezione conferita dai corpi chetonici potrebbe non essere interamente attribuibile allo stress ossidativo. Ad esempio, durante la privazione del glucosio, in un modello di deprivazione del glucosio nei neuroni corticali,? OHB ha stimolato il flusso autofagico e ha prevenuto l'accumulo di autofagosomi, che era associato a una diminuzione della morte neuronale (Camberos-Luna et al., 2016). d-? OHB induce anche le proteine antiossidanti canoniche FOXO3a, SOD, MnSOD e catalasi, prospetticamente attraverso l'inibizione di HDAC (Nagao et al., 2016; Shimazu et al., 2013).
Malattia del fegato grasso non alcolico (NAFLD) e metabolismo del corpo chetone
La NAFLD associata all'obesità e la steatoepatite non alcolica (NASH) sono le cause più comuni di malattia del fegato nei paesi occidentali (Rinella e Sanyal, 2016) e l'insufficienza epatica indotta dalla NASH è uno dei motivi più comuni per il trapianto di fegato. Mentre l'eccessivo stoccaggio di triacilgliceroli negli epatociti> 5% del peso del fegato (NAFL) da solo non causa una funzionalità epatica degenerativa, la progressione a NAFLD nell'uomo è correlata alla resistenza sistemica all'insulina e all'aumento del rischio di diabete di tipo 2 e può contribuire alla patogenesi del malattie cardiovascolari e malattie renali croniche (Fabbrini et al., 2009; Targher et al., 2010; Targher e Byrne, 2013). I meccanismi patogeni di NAFLD e NASH non sono completamente compresi ma includono anomalie del metabolismo degli epatociti, autofagia degli epatociti e stress del reticolo endoplasmatico, funzione delle cellule immunitarie epatiche, infiammazione del tessuto adiposo e mediatori infiammatori sistemici (Fabbrini et al., 2009; Masuoka e Chalasani, 2013 ; Targher et al., 2010; Yang et al., 2010). Le perturbazioni del metabolismo di carboidrati, lipidi e aminoacidi si verificano e contribuiscono all'obesità, al diabete e alla NAFLD negli esseri umani e negli organismi modello [rivisto in (Farese et al., 2012; Lin e Accili, 2011; Newgard, 2012; Samuel e Shulman, 2012; Sun e Lazar, 2013)]. Mentre le anomalie degli epatociti nel metabolismo dei lipidi citoplasmatici sono comunemente osservate nella NAFLD (Fabbrini et al., 2010b), il ruolo del metabolismo mitocondriale, che governa lo smaltimento ossidativo dei grassi è meno chiaro nella patogenesi della NAFLD. Anomalie del metabolismo mitocondriale si verificano e contribuiscono alla patogenesi NAFLD / NASH (Hyotylainen et al., 2016; Serviddio et al., 2011; Serviddio et al., 2008; Wei et al., 2008). C'è generale (Felig et al., 1974; Iozzo et al., 2010; Koliaki et al., 2015; Satapati et al., 2015; Satapati et al., 2012; Sunny et al., 2011) ma non uniforme ( Koliaki e Roden, 2013; Perry et al., 2016; Rector et al., 2010) consenso sul fatto che, prima dello sviluppo della NASH autentica, l'ossidazione mitocondriale epatica, e in particolare l'ossidazione dei grassi, è aumentata nell'obesità, insulino-resistenza sistemica e NAFLD. È probabile che con il progredire della NAFLD, emerga l'eterogenicità della capacità ossidativa, anche tra i singoli mitocondri, e alla fine la funzione ossidativa venga compromessa (Koliaki et al., 2015; Rector et al., 2010; Satapati et al., 2008; Satapati et al. ., 2012).
La chetogenesi viene spesso utilizzata come proxy per l'ossidazione dei grassi epatici. Le alterazioni della chetogenesi emergono man mano che la NAFLD progredisce nei modelli animali e probabilmente negli esseri umani. Attraverso meccanismi non completamente definiti, l'iperinsulinemia sopprime la chetogenesi, probabilmente contribuendo all'ipoketonemia rispetto ai controlli magri (Bergman et al., 2007; Bickerton et al., 2008; Satapati et al., 2012; Soeters et al., 2009; Sunny et al. , 2011; Vice et al., 2005). Tuttavia, la capacità della circolazione delle concentrazioni corporee di chetoni di predire la NAFLD è controversa (Mnnist et al., 2015; Sanyal et al., 2001). Robusti metodi spettroscopici di risonanza magnetica quantitativa in modelli animali hanno rivelato un aumento del tasso di turnover chetonico con moderata insulino-resistenza, ma tassi ridotti erano evidenti con una più grave resistenza all'insulina (Satapati et al., 2012; Sunny et al., 2010). Negli esseri umani obesi con fegato grasso, il tasso chetogenico è normale (Bickerton et al., 2008; Sunny et al., 2011), e quindi, i tassi di chetogenesi sono diminuiti rispetto all'aumento del carico di acidi grassi all'interno degli epatociti. Di conseguenza, l'acetil-CoA derivato dall'ossidazione può essere diretto all'ossidazione terminale nel ciclo TCA, aumentando l'ossidazione terminale, la gluconeogenesi guidata da fosfoenolpiruvato tramite anaplerosi / cataplerosi e stress ossidativo. L'acetil-CoA può anche essere esportato dai mitocondri come citrato, un substrato precursore per la lipogenesi (Fig.4) (Satapati et al., 2015; Satapati et al., 2012; Solinas et al., 2015). Mentre la chetogenesi diventa meno reattiva all'insulina o al digiuno con obesità prolungata (Satapati et al., 2012), i meccanismi sottostanti e le conseguenze a valle di ciò rimangono non completamente compresi. Prove recenti indicano che mTORC1 sopprime la chetogenesi in un modo che potrebbe essere a valle della segnalazione dell'insulina (Kucejova et al., 2016), che è concorde con le osservazioni secondo cui mTORC1 inibisce l'induzione di Hmgcs2 mediata da PPAR? (Sengupta et al., 2010) ( vedere anche il regolamento di HMGCS2 e SCOT / OXCT1).
ï ¿½
Le osservazioni preliminari del nostro gruppo suggeriscono conseguenze epatiche avverse dell'insufficienza chetogenica (Cotter et al., 2014). Per verificare l'ipotesi che la chetogenesi alterata, anche in stati ricchi di carboidrati e quindi `` non chetogenici '', contribuisca al metabolismo del glucosio anormale e provochi la steatoepatite, abbiamo generato un modello murino di marcata insufficienza chetogenica mediante somministrazione di oligonucleotidi antisenso (ASO) mirati a Hmgcs2. La perdita di HMGCS2 nei topi adulti nutriti con cibo a basso contenuto di grassi standard ha causato una lieve iperglicemia e una produzione notevolmente aumentata di centinaia di metaboliti epatici, una suite dei quali ha fortemente suggerito l'attivazione della lipogenesi. L'alimentazione con dieta ad alto contenuto di grassi di topi con chetogenesi insufficiente ha provocato un esteso danno epatocitario e infiammazione. Questi risultati supportano l'ipotesi centrale che (i) la chetogenesi non sia una via di trabocco passivo ma piuttosto un nodo dinamico nell'omeostasi fisiologica epatica e integrata, e (ii) l'aumento chetogenico prudente per mitigare NAFLD / NASH e il metabolismo epatico disordinato del glucosio merita di essere esplorato .
In che modo la chetogenesi alterata può contribuire al danno epatico e all'alterazione dell'omeostasi del glucosio? La prima considerazione è se il colpevole sia la carenza di flusso chetogenico o gli stessi chetoni. Un recente rapporto suggerisce che i corpi chetonici possono mitigare il danno epatico indotto dallo stress ossidativo in risposta agli acidi grassi polinsaturi n-3 (Pawlak et al., 2015). Ricorda che a causa della mancanza di espressione di SCOT negli epatociti, i corpi chetonici non sono ossidati, ma possono contribuire alla lipogenesi e svolgere una varietà di ruoli di segnalazione indipendenti dalla loro ossidazione (vedi anche Destini metabolici non ossidativi dei corpi chetonici e? OHB come un mediatore di segnalazione). È anche possibile che i corpi chetonici derivati dagli epatociti possano servire come segnale e / o metabolita per i tipi di cellule adiacenti all'interno dell'acino epatico, comprese le cellule stellate e i macrofagi delle cellule di Kupffer. Mentre la letteratura limitata disponibile suggerisce che i macrofagi non sono in grado di ossidare i corpi chetonici, questo è stato misurato solo utilizzando metodologie classiche e solo nei macrofagi peritoneali (Newsholme et al., 1986; Newsholme et al., 1987), indicando che un ri- la valutazione è appropriata data l'abbondante espressione di SCOT nei macrofagi derivati dal midollo osseo (Youm et al., 2015).
Anche il flusso chetogenico degli epatociti può essere citoprotettivo. Sebbene i meccanismi salutari possano non dipendere dalla chetogenesi di per sé, le diete chetogeniche a basso contenuto di carboidrati sono state associate al miglioramento della NAFLD (Browning et al., 2011; Foster et al., 2010; Kani et al., 2014; Schugar e Crawford, 2012) . Le nostre osservazioni indicano che la chetogenesi degli epatociti può feedback e regolare il flusso del ciclo TCA, il flusso anaplerotico, la gluconeogenesi derivata dal fosfoenolpiruvato (Cotter et al., 2014) e persino il turnover del glicogeno. La compromissione chetogenica indirizza l'acetil-CoA ad aumentare il flusso di TCA, che nel fegato è stato collegato a un aumento del danno mediato da ROS (Satapati et al., 2015; Satapati et al., 2012); forza la diversione del carbonio in specie lipidiche sintetizzate de novo che potrebbero rivelarsi citotossiche; e previene la riossidazione del NADH a NAD + (Cotter et al., 2014) (Fig. 4). Presi insieme, sono necessari esperimenti futuri per affrontare i meccanismi attraverso i quali l'insufficienza chetogenica relativa può diventare disadattativa, contribuire all'iperglicemia, provocare steatoepatite e se questi meccanismi sono operativi nella NAFLD / NASH umana. Poiché l'evidenza epidemiologica suggerisce una chetogenesi compromessa durante la progressione della steatoepatite (Embade et al., 2016; Marinou et al., 2011; Mnnisté et al., 2015; Pramfalk et al., 2015; Safaei et al., 2016) le terapie che aumentano la chetogenesi epatica potrebbero rivelarsi salutari (Degirolamo et al., 2016; Honda et al., 2016).
Corpi chetonici e insufficienza cardiaca (HF)
Con un tasso metabolico superiore a 400 kcal / kg / giorno e un turnover di 6 35 kg ATP / giorno, il cuore è l'organo con il più alto dispendio energetico e richiesta ossidativa (Ashrafian et al., 2007; Wang et al., 2010b). La stragrande maggioranza del ricambio energetico del miocardio risiede nei mitocondri e il 70% di questa fornitura proviene dalla FAO. Il cuore è onnivoro e flessibile in condizioni normali, ma il cuore che si rimodella patologicamente (p. Es., A causa di ipertensione o infarto miocardico) e il cuore diabetico diventano metabolicamente inflessibili (Balasse e Fery, 1989; BING, 1954; Fukao et al., 2004 ; Lopaschuk et al., 2010; Taegtmeyer et al., 1980; Taegtmeyer et al., 2002; Young et al., 2002). Infatti, le anomalie geneticamente programmate del metabolismo del carburante cardiaco nei modelli murini provocano cardiomiopatia (Carley et al., 2014; Neubauer, 2007). In condizioni fisiologiche i cuori normali ossidano i corpi chetonici in proporzione alla loro consegna, a scapito dell'ossidazione degli acidi grassi e del glucosio, e il miocardio è il più alto consumatore di corpi chetonici per unità di massa (BING, 1954; Crawford et al., 2009; GARLAND et al. ., 1962; Hasselbaink et al., 2003; Jeffrey et al., 1995; Pelletier et al., 2007; Tardif et al., 2001; Yan et al., 2009). Rispetto all'ossidazione degli acidi grassi, i corpi chetonici sono più efficienti dal punto di vista energetico, producendo più energia disponibile per la sintesi di ATP per molecola di ossigeno investita (rapporto P / O) (Kashiwaya et al., 2010; Sato et al., 1995; Veech, 2004) . L'ossidazione del corpo chetonico produce anche energia potenzialmente più elevata della FAO, mantenendo l'ubichinone ossidato, il che aumenta la capacità redox nella catena di trasporto degli elettroni e rende disponibile più energia per sintetizzare l'ATP (Sato et al., 1995; Veech, 2004). L'ossidazione dei corpi chetonici può anche ridurre la produzione di ROS e quindi lo stress ossidativo (Veech, 2004).
Studi preliminari interventistici e osservazionali indicano un potenziale ruolo salutare dei corpi chetonici nel cuore. Nel contesto sperimentale dell'ischemia / riperfusione, i corpi chetonici hanno conferito potenziali effetti cardioprotettivi (Al-Zaid et al., 2007; Wang et al., 2008), probabilmente a causa dell'aumento dell'abbondanza mitocondriale nel cuore o dell'up-regolazione della fosforilazione ossidativa cruciale mediatori (Snorek et al., 2012; Zou et al., 2002). Studi recenti indicano che l'utilizzazione del corpo chetone è aumentata nel cuore di topi (Aubert et al., 2016) e nell'uomo (Bedi et al., 2016), supportando osservazioni precedenti nell'uomo (BING, 1954; Fukao et al., 2000; Janardhan et al., 2011; Longo et al., 2004; Rudolph e Schinz, 1973; Tildon e Cornblath, 1972). Le concentrazioni circolanti del corpo chetonico sono aumentate nei pazienti con insufficienza cardiaca, in proporzione diretta alle pressioni di riempimento, osservazioni il cui meccanismo e significato rimane sconosciuto (Kupari et al., 1995; Lommi et al., 1996; Lommi et al., 1997; Neely et al ., 1972), ma i topi con deficit di SCOT selettivo nei cardiomiociti mostrano un rimodellamento ventricolare patologico accelerato e firme ROS in risposta al danno da sovraccarico pressorio indotto chirurgicamente (Schugar et al., 2014).
Recenti osservazioni intriganti nella terapia del diabete hanno rivelato un potenziale legame tra il metabolismo chetone miocardico e il rimodellamento ventricolare patologico (Fig. 5). L'inibizione del co-trasportatore renale prossimale tubulare di sodio / glucosio 2 (SGLT2i) aumenta le concentrazioni circolanti di corpi chetonici negli esseri umani (Ferrannini et al., 2016a; Inagaki et al., 2015) e topi (Suzuki et al., 2014) aumentati chetogenesi epatica (Ferrannini et al., 2014; Ferrannini et al., 2016a; Katz e Leiter, 2015; Mudaliar et al., 2015). Sorprendentemente, almeno uno di questi agenti ha ridotto il ricovero in HF (ad esempio, come rivelato dallo studio EMPA-REG OUTCOME) e ha migliorato la mortalità cardiovascolare (Fitchett et al., 2016; Sonesson et al., 2016; Wu et al., 2016a ; Zinman et al., 2015). Mentre i meccanismi di guida dietro benefici esiti di HF a SGLT2i rimangono attivamente dibattuti, il beneficio di sopravvivenza è probabilmente multifattoriale, includendo in modo prospettico la chetosi ma anche effetti salutari su peso, pressione sanguigna, glucosio e livelli di acido urico, rigidità arteriosa, sistema nervoso simpatico, osmotico diuresi / riduzione del volume plasmatico e aumento dell'ematocrito (Raz e Cahn, 2016; Vallon e Thomson, 2016). Nel complesso, la nozione che aumentare la chetonemia terapeuticamente sia nei pazienti con scompenso cardiaco, sia in quelli ad alto rischio di sviluppare HF, rimane controversa ma è oggetto di studio attivo in studi pre-clinici e clinici (Ferrannini et al., 2016b; Kolwicz et al., 2016; Lopaschuk e Verma, 2016; Mudaliar et al., 2016; Taegtmeyer, 2016).
ï ¿½
Corpi chetonici in biologia del cancro
I collegamenti tra corpi chetonici e cancro stanno rapidamente emergendo, ma gli studi su entrambi i modelli animali e sull'uomo hanno portato a conclusioni diverse. Poiché il metabolismo chetone è dinamico e reattivo allo stato nutritivo, è interessante per perseguire legami biologici con il cancro a causa del potenziale di terapie nutrizionali guidate con precisione. Le cellule tumorali sono sottoposte a riprogrammazione metabolica per mantenere una rapida proliferazione e crescita cellulare (DeNicola e Cantley, 2015, Pavlova e Thompson, 2016). Il classico effetto Warburg nel metabolismo delle cellule cancerogene deriva dal ruolo dominante della glicolisi e della fermentazione dell'acido lattico per trasferire energia e compensare una minore dipendenza dalla fosforilazione ossidativa e dalla limitata respirazione mitocondriale (De Feyter et al., 2016; Grabacka et al., 2016; Kang et al., 2015; Poff et al., 2014; Shukla et al., 2014). Il carbonio glucosio è principalmente diretto attraverso la glicolisi, la via del pentoso fosfato e la lipogenesi, che insieme forniscono gli intermedi necessari per l'espansione della biomassa tumorale (Grabacka et al., 2016; Shukla et al., 2014; Yoshii et al., 2015). L'adattamento delle cellule tumorali alla deprivazione di glucosio avviene attraverso la capacità di sfruttare fonti di carburante alternative, tra cui acetato, glutammina e aspartato (Jaworski et al., 2016; Sullivan et al., 2015). Ad esempio, l'accesso limitato al piruvato rivela la capacità delle cellule tumorali di convertire la glutammina in acetil-CoA mediante carbossilazione, mantenendo sia i bisogni energetici che anabolici (Yang et al., 2014). Un interessante adattamento delle cellule cancerose è l'utilizzo dell'acetato come combustibile (Comerford et al., 2014; Jaworski et al., 2016; Mashimo et al., 2014; Wright e Simone, 2016; Yoshii et al., 2015). L'acetato è anche un substrato per la lipogenesi, che è fondamentale per la proliferazione delle cellule tumorali, e il guadagno di questo condotto lipogenico è associato alla sopravvivenza dei pazienti più breve e al carico tumorale maggiore (Comerford et al., 2014; Mashimo et al., 2014; Yoshii et al ., 2015).
Le cellule non cancerose spostano facilmente la loro fonte di energia dal glucosio ai corpi chetonici durante la privazione del glucosio. Questa plasticità può essere più variabile tra i tipi di cellule tumorali, ma i tumori cerebrali impiantati in vivo hanno ossidato [2,4-13C2] -? OHB in misura simile al tessuto cerebrale circostante (De Feyter et al., 2016). I modelli con effetto Warburg inverso o metabolismo a due compartimenti ipotizzano che le cellule tumorali inducano la produzione di? OHB nei fibroblasti adiacenti, fornendo il fabbisogno energetico delle cellule tumorali (Bonuccelli et al., 2010; Martinez-Outschoorn et al., 2012) . Nel fegato, uno spostamento degli epatociti dalla chetogenesi all'ossidazione dei chetoni nelle cellule di carcinoma epatocellulare (epatoma) è coerente con l'attivazione delle attività BDH1 e SCOT osservate in due linee cellulari di epatoma (Zhang et al., 1989). In effetti, le cellule dell'epatoma esprimono OXCT1 e BDH1 e ossidano i chetoni, ma solo quando il siero muore di fame (Huang et al., 2016). In alternativa, è stata proposta anche la chetogenesi delle cellule tumorali. Cambiamenti dinamici nell'espressione genica chetogenica si manifestano durante la trasformazione cancerosa dell'epitelio del colon, un tipo di cellula che normalmente esprime HMGCS2, e un recente rapporto ha suggerito che HMGCS2 può essere un marker prognostico di prognosi sfavorevole nei carcinomi a cellule squamose e del colon-retto (Camarero et al., 2006; Chen et al., 2016). Resta da determinare se questa associazione richieda o coinvolga la chetogenesi o una funzione di luce lunare di HMGCS2. Al contrario, la produzione apparente di? OHB da parte delle cellule di melanoma e glioblastoma, stimolata dal PPAR? fenofibrato agonista, è stato associato all'arresto della crescita (Grabacka et al., 2016). Sono necessari ulteriori studi per caratterizzare i ruoli dell'espressione di HMGCS2 / SCOT, della chetogenesi e dell'ossidazione dei chetoni nelle cellule tumorali.
Al di là del regno del metabolismo del carburante, i chetoni sono stati recentemente coinvolti nella biologia delle cellule tumorali tramite un meccanismo di segnalazione. L'analisi del melanoma BRAF-V600E + ha indicato l'induzione di HMGCL dipendente da OCT1 in modo oncogenico BRAF-dipendente (Kang et al., 2015). L'aumento di HMGCL è stato correlato con una maggiore concentrazione di AcAc cellulare, che a sua volta ha migliorato l'interazione BRAFV600E-MEK1, amplificando la segnalazione MEK-ERK in un loop feed-forward che guida la proliferazione e la crescita delle cellule tumorali. Queste osservazioni sollevano l'interessante questione della chetogenesi extraepatica prospettica che quindi supporta un meccanismo di segnalazione (vedi anche? OHB come mediatore di segnalazione e Controversies in extraepatic chetogenesis). È anche importante considerare gli effetti indipendenti di AcAc, d-? OHB e l-? OHB sul metabolismo del cancro e, quando si considera l'HMGCL, anche il catabolismo della leucina può essere squilibrato.
Gli effetti delle diete chetogeniche (vedere anche Uso terapeutico della dieta chetogenica e dei corpi chetonici esogeni) nei modelli animali di cancro sono vari (De Feyter et al., 2016; Klement et al., 2016; Meidenbauer et al., 2015; Poff et al. ., 2014; Seyfried et al., 2011; Shukla et al., 2014). Mentre le associazioni epidemiologiche tra obesità, cancro e diete chetogeniche sono dibattute (Liskiewicz et al., 2016; Wright e Simone, 2016), una meta-analisi che utilizza diete chetogeniche in modelli animali e in studi sull'uomo ha suggerito un impatto salutare sulla sopravvivenza, con benefici prospetticamente collegati all'entità della chetosi, al momento dell'inizio della dieta e alla localizzazione del tumore (Klement et al., 2016; Woolf et al., 2016). Il trattamento delle cellule tumorali del pancreas con corpi chetonici (d-? OHB o AcAc) ha inibito la crescita, la proliferazione e la glicolisi e una dieta chetogenica (81% kcal di grassi, 18% di proteine, 1% di carboidrati) ha ridotto in vivo il peso del tumore, la glicemia e aumento del peso muscolare e corporeo negli animali con cancro impiantato (Shukla et al., 2014). Risultati simili sono stati osservati utilizzando un modello di cellule di glioblastoma metastatico in topi che hanno ricevuto l'integrazione di chetoni nella dieta (Poff et al., 2014). Al contrario, una dieta chetogenica (91% kcal di grassi, 9% di proteine) aumenta la concentrazione di OHB circolante e diminuisce la glicemia, ma non ha avuto alcun impatto sul volume del tumore o sulla durata di sopravvivenza nei ratti portatori di glioma (De Feyter et al., 2016). Un indice di glucosio chetone è stato proposto come indicatore clinico che migliora la gestione metabolica della terapia del cancro al cervello indotta dalla dieta chetogenica nell'uomo e nei topi (Meidenbauer et al., 2015). Presi insieme, i ruoli del metabolismo dei corpi chetonici e dei corpi chetonici nella biologia del cancro sono allettanti perché ciascuno di essi pone opzioni terapeutiche trattabili, ma gli aspetti fondamentali rimangono da chiarire, con chiare influenze che emergono da una matrice di variabili, comprese (i) differenze tra chetoni esogeni corpi contro dieta chetogenica, (ii) tipo di cellula cancerosa, polimorfismi genomici, grado e stadio; e (iii) tempi e durata dell'esposizione allo stato chetotico.
La chetogenesi è creata da corpi chetonici attraverso la scissione di acidi grassi e amminoacidi chetogenici. Questo processo biochimico fornisce energia a vari organi, in particolare il cervello, in circostanze di digiuno come risposta ad una non disponibilità di glucosio nel sangue. I corpi chetonici sono prodotti principalmente nei mitocondri delle cellule del fegato. Mentre altre cellule sono in grado di effettuare la chetogenesi, non sono così efficaci nel farlo come le cellule del fegato. Poiché la chetogenesi si verifica nei mitocondri, i suoi processi sono regolati indipendentemente. Dr. Alex Jimenez DC, CCST Insight
Applicazione terapeutica della dieta chetogenica e dei corpi chetonici esogeni
Le applicazioni delle diete chetogeniche e dei corpi chetonici come strumenti terapeutici sono sorte anche in contesti non cancerosi tra cui l'obesità e NAFLD / NASH (Browning et al., 2011; Foster et al., 2010; Schugar e Crawford, 2012); insufficienza cardiaca (Huynh, 2016; Kolwicz et al., 2016; Taegtmeyer, 2016); malattie neurologiche e neurodegenerative (Martin et al., 2016; McNally e Hartman, 2012; Rho, 2015; Rogawski et al., 2016; Yang e Cheng, 2010; Yao et al., 2011); errori congeniti del metabolismo (Scholl-B rgi et al, 2015); e la prestazione fisica (Cox et al., 2016). L'efficacia delle diete chetogeniche è stata particolarmente apprezzata nella terapia delle crisi epilettiche, in particolare nei pazienti resistenti ai farmaci. La maggior parte degli studi ha valutato le diete chetogeniche nei pazienti pediatrici e rivela una riduzione fino al 50% circa della frequenza delle crisi dopo 3 mesi, con una migliore efficacia in sindromi selezionate (Wu et al., 2016b). L'esperienza è più limitata nell'epilessia degli adulti, ma una riduzione simile è evidente, con una migliore risposta nei pazienti con epilessia generalizzata sintomatica (Nei et al., 2014). I meccanismi anticonvulsivanti sottostanti rimangono poco chiari, sebbene le ipotesi postulate includano un ridotto utilizzo / glicolisi del glucosio, trasporto riprogrammato del glutammato, impatto indiretto sul canale del potassio ATP-sensibile o sul recettore dell'adenosina A1, alterazione dell'espressione dell'isoforma del canale del sodio o effetti sugli ormoni circolanti inclusa la leptina ( Lambrechts et al., 2016; Lin et al., 2017; Lutas e Yellen, 2013). Non è chiaro se l'effetto anticonvulsivante sia principalmente attribuibile ai corpi chetonici o alle conseguenze metaboliche a cascata delle diete a basso contenuto di carboidrati. Tuttavia, gli esteri chetonici (vedi sotto) sembrano elevare la soglia convulsiva nei modelli animali di convulsioni provocate (Ciarlone et al., 2016; D'Agostino et al., 2013; Viggiano et al., 2015).
Le diete atkins e chetogeniche, a basso contenuto di carboidrati sono spesso considerate sgradevoli e possono causare stitichezza, iperuricemia, ipocalcemia, ipomagnesiemia, portare a nefrolitiasi, chetoacidosi, causare iperglicemia e aumentare il colesterolo circolante e le concentrazioni di acidi grassi liberi (Bisschop et al., 2001 ; Kossoff e Hartman, 2012; Kwiterovich et al., 2003; Suzuki et al., 2002). Per questi motivi, l'aderenza a lungo termine pone delle sfide. Gli studi sui roditori usano comunemente una distribuzione distintiva dei macronutrienti (94% kcal di grasso, 1% di carboidrati kcal, 5% di kcal di proteine, Bio-Serv F3666), che provoca una chetosi robusta. Tuttavia, l'aumento del contenuto proteico, anche a 10% kcal, diminuisce sostanzialmente la chetosi e la restrizione della proteina 5% kcal conferisce confondenti effetti metabolici e fisiologici. Questa formulazione dietetica è anche colina esaurita, un'altra variabile che influenza la suscettibilità al danno epatico e persino la chetogenesi (Garbow et al., 2011; Jornayvaz et al., 2010; Kennedy et al., 2007; Pissios et al., 2013; Schugar et al., 2013). Gli effetti del consumo a lungo termine di diete chetogeniche nei topi rimangono incompleti, ma studi recenti sui topi hanno rivelato una sopravvivenza normale e l'assenza di marcatori di danno epatico nei topi su diete chetogeniche nel corso della loro vita, sebbene il metabolismo degli aminoacidi, il dispendio energetico e la segnalazione di insulina sono stati notevolmente riprogrammati (Douris et al., 2015).
Meccanismi che aumentano la chetosi attraverso meccanismi alternativi alle diete chetogeniche includono l'uso di precursori del corpo chetone ingestibili. La somministrazione di corpi chetonici esogeni potrebbe creare uno stato fisiologico unico non riscontrato nella fisiologia normale, poiché le concentrazioni di glucosio e insulina circolanti sono relativamente normali, mentre le cellule potrebbero risparmiare l'assorbimento e l'utilizzo di glucosio. Gli stessi corpi chetonici hanno una breve emivita e l'ingestione o l'infusione di sale di sodio-OHB per ottenere la chetosi terapeutica provoca un carico di sodio indesiderato. L'R / S-1,3-butandiolo è un dialcool non tossico che viene facilmente ossidato nel fegato per produrre d / l-? OHB (Desrochers et al., 1992). In contesti sperimentali distinti, questa dose è stata somministrata quotidianamente a topi o ratti per un massimo di sette settimane, producendo concentrazioni di? OHB circolanti fino a 5 mM entro 2 ore dalla somministrazione, che è stabile per almeno altre 3 ore (D ' Agostino et al., 2013). La soppressione parziale dell'assunzione di cibo è stata osservata nei roditori dati R / S-1,3-butandiolo (Carpenter and Grossman, 1983). Inoltre, tre esteri chetonici (KE) chimicamente distinti, (i) monoestere di R-1,3-butandiolo e d-? OHB (R-3-idrossibutil R-? OHB); (ii) gliceril-tris-? OHB; e (iii) R, S-1,3-butandiolo acetoacetato diestere, sono stati anche ampiamente studiati (Brunengraber, 1997; Clarke et al., 2012a; Clarke et al., 2012b; Desrochers et al., 1995a; Desrochers et al. ., 1995b; Kashiwaya et al., 2010). Un vantaggio intrinseco del primo è che 2 moli di d-? OHB fisiologico vengono prodotte per mole di KE, in seguito all'idrolisi dell'esterasi nell'intestino o nel fegato. La sicurezza, la farmacocinetica e la tolleranza sono state studiate più ampiamente negli esseri umani che ingeriscono R-3-idrossibutil R-? OHB, a dosi fino a 714 mg / kg, producendo concentrazioni circolanti di d-? OHB fino a 6 mM (Clarke et al., 2012a; Cox et al., 2016; Kemper et al., 2015; Shivva et al., 2016). Nei roditori, questo KE riduce l'apporto calorico e il colesterolo totale plasmatico, stimola il tessuto adiposo bruno e migliora la resistenza all'insulina (Kashiwaya et al., 2010; Kemper et al., 2015; Veech, 2013). Recenti scoperte indicano che durante l'esercizio in atleti allenati, l'ingestione di R-3-idrossibutil R-? OHB ha diminuito la glicolisi dei muscoli scheletrici e le concentrazioni di lattato plasmatico, aumentato l'ossidazione intramuscolare del triacilglicerolo e conservato il contenuto di glicogeno muscolare, anche quando i carboidrati ingeriti hanno stimolato la secrezione di insulina ( Cox et al., 2016). È necessario un ulteriore sviluppo di questi risultati intriganti, poiché il miglioramento delle prestazioni degli esercizi di resistenza è stato determinato principalmente da una robusta risposta al KE nei soggetti 2 / 8. Tuttavia, questi risultati supportano studi classici che indicano una preferenza per l'ossidazione chetone su altri substrati (GARLAND et al., 1962; Hasselbaink et al., 2003; Stanley et al., 2003; Valente-Silva et al., 2015), incluso durante l'esercizio, e che gli atleti addestrati possono essere più predisposti ad utilizzare i chetoni (Johnson et al., 1969a; Johnson e Walton, 1972; Winder et al., 1974; Winder et al., 1975). Infine, restano da determinare i meccanismi che potrebbero supportare una migliore prestazione fisica a parità di apporto calorico (differenzialmente distribuito tra macronutrienti) e tassi di consumo di ossigeno uguali.
Prospettiva futura
Una volta ampiamente stigmatizzata come una via di trabocco in grado di accumulare emissioni tossiche dalla combustione dei grassi in stati di ristrettezze di carboidrati (il paradigma `` chetotossico ''), recenti osservazioni supportano l'idea che il metabolismo corporeo chetone svolga ruoli salutari anche negli stati carichi di carboidrati, aprendo una `` chetoormetica '' ipotesi. Mentre i facili approcci nutrizionali e farmacologici per manipolare il metabolismo dei chetoni lo rendono un bersaglio terapeutico attraente, gli esperimenti proposti in modo aggressivo ma prudenti rimangono sia nei laboratori di ricerca di base che in quelli traslazionali. Sono emerse esigenze insoddisfatte nell'ambito della definizione del ruolo di sfruttare il metabolismo dei chetoni nell'insufficienza cardiaca, nell'obesità, nella NAFLD / NASH, nel diabete di tipo 2 e nel cancro. La portata e l'impatto dei ruoli di segnalazione `` non canonici '' dei corpi chetonici, inclusa la regolazione dei PTM che probabilmente si alimentano avanti e indietro nelle vie metaboliche e di segnalazione, richiedono un'esplorazione più approfondita. Infine, la chetogenesi extraepatica potrebbe aprire intriganti meccanismi di segnalazione paracrina e autocrina e opportunità per influenzare il co-metabolismo all'interno del sistema nervoso e dei tumori per raggiungere scopi terapeutici.
In conclusione, i corpi chetonici vengono creati dal fegato per essere utilizzati come fonte di energia quando non c'è abbastanza glucosio prontamente disponibile nel corpo umano. La chetogenesi si verifica quando ci sono bassi livelli di glucosio nel sangue, in particolare dopo che le altre riserve di carboidrati cellulari sono state esaurite. Lo scopo dell'articolo sopra era quello di discutere i ruoli multidimensionali dei corpi chetonici nel metabolismo del carburante, nella segnalazione e nella terapia. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
Discussione aggiuntiva sull'argomento: Dolore alla schiena acuto
Mal di schienaè una delle cause più diffuse di disabilità e di giornate di lavoro perse in tutto il mondo. Il dolore alla schiena si attribuisce al secondo motivo più comune per le visite presso l'ambulatorio medico, superato solo dalle infezioni delle vie respiratorie superiori. Circa l'80% della popolazione sperimenterà dolore alla schiena almeno una volta nella vita. La colonna vertebrale è una struttura complessa composta da ossa, articolazioni, legamenti e muscoli, tra gli altri tessuti molli. Lesioni e / o condizioni aggravate, come dischi erniciati, può eventualmente portare a sintomi di mal di schiena. Gli infortuni sportivi o gli incidenti automobilistici sono spesso la causa più frequente di mal di schiena, tuttavia, a volte il più semplice dei movimenti può avere risultati dolorosi. Fortunatamente, opzioni di trattamento alternative, come la cura chiropratica, possono aiutare ad alleviare il mal di schiena attraverso l'uso di aggiustamenti spinali e manipolazioni manuali, migliorando in definitiva il sollievo dal dolore.
I eritroide nucleare 2 correlato al fattore 2 signaling pathway, meglio noto come Nrf2, è un meccanismo protettivo che funziona come un "regolatore principale" della risposta antiossidante del corpo umano. Nrf2 rileva i livelli di stress ossidativo all'interno delle cellule e innesca meccanismi antiossidanti protettivi. Mentre l'attivazione di Nrf2 può avere molti vantaggi, la "sovraespressione" di Nrf2 può avere diversi rischi.
Sembra che un livello equilibrato di NRF2 sia essenziale per prevenire lo sviluppo generale di una varietà di malattie oltre al miglioramento generale di questi problemi di salute. Tuttavia, NRF2 può anche causare complicazioni. La causa principale della "sovraespressione" di NRF2 è dovuta a una mutazione genetica o ad un'esposizione cronica continua a uno stress chimico o ossidativo, tra gli altri. Di seguito, discuteremo gli aspetti negativi della sovraespressione di Nrf2 e mostreremo i suoi meccanismi d'azione all'interno del corpo umano.
Cancro
Studi di ricerca hanno scoperto che i topi che non esprimono NRF2 sono più inclini a sviluppare il cancro in risposta alla stimolazione fisica e chimica. Studi di ricerca simili, tuttavia, hanno dimostrato che l'eccessiva attivazione di NRF2, o anche l'inattivazione di KEAP1, può provocare l'esacerbazione di alcuni tumori, in particolare se tali percorsi sono stati interrotti. L'iperattività NRF2 può verificarsi attraverso il fumo, dove si ritiene che l'attivazione continua di NRF2 sia la causa del cancro ai polmoni nei fumatori. La sovraespressione di Nrf2 potrebbe impedire alle cellule cancerose di autodistruggersi, mentre l'attivazione intermittente di NRF2 può impedire alle cellule cancerose di innescare l'induzione di tossine.
Inoltre, poiché la sovraespressione di NRF2 aumenta la capacità antiossidante del corpo umano di funzionare oltre l'omeostasi redox, questo aumenta la divisione cellulare e genera un modello innaturale di metilazione del DNA e degli istoni. In ultima analisi, ciò può rendere la chemioterapia e la radioterapia meno efficaci contro il cancro. Pertanto, limitare l'attivazione di NRF2 con sostanze come DIM, luteolina, Zi Cao o salinomicina potrebbe essere l'ideale per i pazienti con cancro, sebbene l'iperattivazione di Nrf2 non dovrebbe essere considerata l'unica causa di cancro. Le carenze nutritive possono influenzare i geni, incluso NRF2. Questo potrebbe essere un modo per capire come le carenze contribuiscono ai tumori.
Fegato
L'overactivation di Nrf2, può anche influenzare la funzione di organi specifici nel corpo umano. La sovraespressione di NRF2 può infine bloccare la produzione del fattore di crescita insulino-1, o IGF-1, dal fegato, che è essenziale per la rigenerazione del fegato.
Cuore
Mentre la sovraespressione acuta di Nrf2 può avere i suoi benefici, la sovraespressione continua di NRF2 può causare effetti dannosi a lungo termine sul cuore, come la cardiomiopatia. L'espressione di NRF2 può essere aumentata attraverso alti livelli di colesterolo o l'attivazione di HO-1. Si ritiene che questo sia il motivo per cui livelli cronici elevati di colesterolo potrebbero causare problemi di salute cardiovascolare.
Vitiligine
È stato anche dimostrato che la sovraespressione di NRF2 inibisce la capacità di repigment in vitiligine in quanto potrebbe ostruire la tirosina, o TYR, azione essenziale per la ripigmentazione attraverso la melaninogenesi. Studi di ricerca hanno dimostrato che questo processo può essere uno dei motivi principali per cui le persone con vitiligine non sembrano attivare Nrf2 con la stessa efficacia delle persone senza vitiligine.
Perché NRF2 potrebbe non funzionare correttamente
ormesi
NRF2 deve essere attivato ormeticamente per poter sfruttare i suoi benefici. In altre parole, Nrf2 non dovrebbe attivarsi ogni minuto o ogni giorno, quindi è una buona idea prendersi delle pause, ad esempio, 5 giorni su 5 giorni liberi o ogni altro giorno. NRF2 deve anche raggiungere una soglia specifica per innescare la sua risposta ormetica, dove un piccolo fattore di stress potrebbe non essere sufficiente per innescarlo.
DJ-1 Ossidazione
Deglycase proteico DJ-1, o semplicemente DJ-1, chiamato anche la proteina della malattia di Parkinson, o PARK7, è un regolatore principale e rivelatore dello stato redox nel corpo umano. DJ-1 è essenziale per regolare quanto a lungo NRF2 può svolgere la sua funzione e produrre una risposta antiossidante. Nel caso in cui DJ-1 si ossidi, le cellule renderanno la proteina DJ-1 meno accessibile.
Questo processo induce l'attivazione di NRF2 a scadere troppo velocemente poiché DJ-1 è fondamentale per mantenere livelli bilanciati di NRF2 e impedire che vengano scomposti nella cella. Nel caso in cui la proteina DJ-1 sia inesistente o perossigenata, l'espressione di NRF2 sarà probabilmente minima, anche usando DIM o attivatori alternativi di NRF2. L'espressione di DJ-1 è indispensabile per ripristinare l'azione NRF2 compromessa.
Malattie croniche
Se hai una malattia cronica, tra cui CIRS, infezioni croniche / disbiosi / SIBO o accumulo di metalli pesanti, come il mercurio e / o quello dai canali radicolari, questi possono ostruire i sistemi di NRF2 e la disintossicazione di fase due. Piuttosto che lo stress ossidativo che trasforma NRF2 in un antiossidante, NRF2 non si innescherà e lo stress ossidativo può rimanere nella cellula e causare danni, il che significa che non c'è risposta antiossidante. Questo è un motivo significativo per cui molte persone con CIRS hanno diverse sensibilità e raggiungono numerosi fattori. Alcune persone credono di avere una risposta herx, tuttavia, questa reazione potrebbe solo danneggiare le cellule più lontane.
Il trattamento della malattia cronica, tuttavia, consentirà al fegato di scaricare le tossine nella bile, sviluppando gradualmente la risposta ormetica dell'attivazione di NRF2. Se la bile rimane tossica e non viene escreta dal corpo umano, riattiverà lo stress ossidativo di NRF2 e ti farà sentire peggio una volta riassorbito dal tratto gastrointestinale, o gastrointestinale. Ad esempio, l'ocratossina A può bloccare NRF2. Oltre a trattare il problema, gli inibitori dell'istone deacetilasi possono bloccare la reazione ossidativa da una serie di fattori che innescano l'attivazione di NRF2, ma potrebbero anche impedire che NRF2 si inneschi normalmente, cosa che alla fine potrebbe non servire al suo scopo.
Disregolazione dell'olio di pesce
I colinergici sono sostanze che aumentano l'acetilcolina, o ACh e colina nel cervello attraverso l'aumento di ACh, in particolare quando inibiscono la rottura di ACh. I pazienti con CIRS hanno spesso problemi con la disregolazione dei livelli di acetilcolina nel corpo umano, specialmente nel cervello. L'olio di pesce innesca NRF2, attivando il suo meccanismo protettivo antiossidante all'interno delle cellule.
Le persone con malattie croniche potrebbero avere problemi con lo stress cognitivo e l'eccitotossicità dell'acetilcolina, dall'accumulo di organofosfati, che potrebbero causare l'infiammazione dell'olio di pesce all'interno del corpo umano. La carenza di colina induce inoltre l'attivazione di NRF2. Includere la colina nella dieta (polifenoli, uova, ecc.) Può aiutare a migliorare gli effetti della disregolazione colinergica.
Cosa diminuisce NRF2?
La riduzione della sovraespressione di NRF2 è la migliore per le persone che hanno il cancro, anche se può essere utile per una varietà di altri problemi di salute.
Dieta, integratori e medicinali comuni:
Apigenina (alte dosi)
Brucea javanica
Castagne
EGCG (alte dosi aumentano NRF2)
Fieno greco (Trigonellina)
Hiba (Hinokitiol /? -Thujaplicin)
Dieta ad alto contenuto di sale
Luteolina (sedano, peperone verde, prezzemolo, foglie di perilla e camomilla - dosi più elevate possono aumentare NRF2 - 40 mg / kg luteolina tre volte a settimana)
Metformina (assunzione cronica)
N-acetil-L-cisteina (NAC, bloccando la risposta ossidativa, esp a dosi elevate)
Buccia d'arancia (con flavonoidi polimetossilati)
Quercetina (dosi più elevate possono aumentare NRF2 - 50 mg / kg / d quercetina)
Salinomicina (farmaco)
Retinolo (acido all trans retinoico)
Vitamina C se combinata con la quercetina
Zi Cao (Purple Gromwel ha Shikonin / Alkannin)
Percorsi e altro:
Bach1
Scommessa
biofilm
Brusatol
camptotecina
DNMT
DPP-23
EZH2
Segnalazione del recettore glucocorticoide (desametasone e betametasone pure)
GSK-3? (feedback normativo)
Attivazione HDAC?
alofuginone
Omocisteina (ALCAR può invertire questo omocisteina indurre bassi livelli di NRF2)
IL-24
Keap1
MDA-7
NF? B
Ocratossina A (specie di aspergillus e pencicllium)
Proteina leucemica promielocitica
p38
p53
p97
Recettore alfa dell'acido retinoico
selenite
SYVN1 (Hrd1)
Inibizione di STAT3 (come il Cryptotanshinone)
Testosterone (e propionato di testosterone, sebbene TP per via intranasale possa aumentare NRF2)
Trecator (Ethionamide)
Trx1 (tramite riduzione di Cys151 in Keap1 o di Cys506 nella regione NLS di Nrf2)
Trolox
vorinostat
Deficit di zinco (peggiora nel cervello)
Nrf2 Meccanismo d'azione
Lo stress ossidativo si innesca attraverso CUL3 dove NRF2 di KEAP1, un inibitore negativo, entra successivamente nel nucleo di queste cellule, stimolando la trascrizione delle ARE, trasformando i solfuri in disolfuri e trasformandoli in più geni antiossidanti, portando alla sovraregolazione di antiossidanti, tali come GSH, GPX, GST, SOD, ecc. Il resto di questi può essere visto nella lista qui sotto:
Aumenta l'AKR
Aumenti ARE
Aumenta ATF4
Aumenta Bcl-xL
Aumenta Bcl-2
Aumenta BDNF
Aumenta BRCA1
Aumenta c-giu
Aumenta CAT
Aumenta il cGMP
Aumenta CKIP-1
Aumenta CYP450
Aumenta Cul3
Aumenta GCL
Aumenta GCLC
Aumenta GCLM
Aumenta GCS
Aumenta GPx
Aumenta GR
Aumenta GSH
Aumenta GST
Aumenta HIF1
Aumenta HO-1
Aumenta HQO1
Aumenta HSP70
Aumenta IL-4
Aumenta IL-5
Aumenta IL-10
Aumenta IL-13
Aumenta K6
Aumenta K16
Aumenta K17
Aumenta il mEH
Aumenta Mrp2-5
Aumenta NADPH
Aumenta Notch 1
Aumenta NQO1
Aumenta il PPAR-alfa
Aumenta il prezzo
Aumenta p62
Aumenta Sesn2
Aumenta Slco1b2
Aumenta gli sMafs
Aumenta il SOD
Aumenta Trx
Aumenta Txn (d)
Aumenta UGT1 (A1 / 6)
Aumenta il VEGF
Riduce ADAMTS (4 / 5)
Riduce l'alfa-SMA
Riduce ALT
Riduce AP1
Riduce l'AST
Riduce Bach1
Riduce COX-2
Riduce DNMT
Riduce FASN
Riduce l'FGF
Riduce l'HDAC
Riduce IFN-?
Riduce le IgE
Riduce IGF-1
Riduce IL-1b
Riduce IL-2
Riduce IL-6
Riduce IL-8
Riduce IL-25
Riduce IL-33
Riduce iNOS
Riduce LT
Riduce Keap1
Riduce MCP-1
Riduce MIP-2
Riduce MMP-1
Riduce MMP-2
Riduce MMP-3
Riduce MMP-9
Riduce MMP-13
Riduce NfkB
Riduce NO
Riduce SIRT1
Riduce TGF-b1
Riduce il TNF-alfa
Riduce Tyr
Riduce VCAM-1
Codificato dal gene NFE2L2, NRF2, o fattore 2 relativo all'eritmoide nucleare 2, è un fattore di trascrizione nella cerniera di base della leucina, o bZIP, superfamiglia che utilizza un Cap'n'Collar o struttura CNC.
Promuove gli enzimi nitrici, gli enzimi di biotrasformazione e i trasportatori di efflusso xenobiotici.
È un regolatore essenziale all'induzione dei geni enzimatici antiossidanti e di disintossicazione di fase II, che proteggono le cellule dai danni causati da stress ossidativo e attacchi elettrofili.
Durante le condizioni omeostatiche, Nrf2 è sequestrato nel citosol attraverso il collegamento corporeo del dominio N-terminale di Nrf2, o la proteina associata a Ech Kelch o Keap1, anche chiamata INrf2 o Inibitore di Nrf2, che inibisce l'attivazione di Nrf2.
Può anche essere controllato dalla selenoproteina dei mammiferi thioredoxin reduttasi 1, o TrxR1, che funziona come regolatore negativo.
A seguito della vulnerabilità a fattori di stress elettrofili, Nrf2 si dissocia da Keap1, traslocando nel nucleo, dove viene poi eterodimerizza con una gamma di proteine regolatorie trascrizionali.
Interazioni frequenti comprendono quelle delle autorità di trascrizione Jun e Fos, che possono essere membri della famiglia delle proteine di attivazione dei fattori di trascrizione.
Dopo la dimerizzazione, questi complessi si legano a componenti reattivi antiossidanti / elettrofili ARE / EpRE e attivano la trascrizione, come nel caso del complesso Jun-Nrf2, o sopprimono la trascrizione, proprio come il complesso Fos-Nrf2.
Il posizionamento dell'ARE, che viene attivato o inibito, determinerà quali geni sono controllati trascrizionalmente da queste variabili.
Quando viene attivato ARE:
L'attivazione della sintesi degli antiossidanti è in grado di disintossicare ROS come catalasi, superossido-dismutasi o SOD, GSH-perossidasi, GSH-reduttasi, GSH-transferasi, NADPH-chinone ossidoreduttasi o NQO1, sistema della monoossigenasi Citocromo P450, tioredossina, tioredossina reduttasi e HSP70.
L'attivazione di questa GSH sintasi consente una notevole crescita del grado intracellulare del GSH, che è abbastanza protettivo.
L'aumento di questa sintesi e gradi di enzimi di fase II come UDP-glucuronosiltransferasi, N-acetiltransferasi e sulfotransferasi.
La sovraregolazione di HO-1, che è un recettore veramente protettivo con una potenziale crescita di CO che, in combinazione con NO, consente la vasodilatazione delle cellule ischemiche.
Riduzione del sovraccarico di ferro attraverso l'elevata ferritina e la bilirubina come antiossidante lipofilo. Sia le proteine di fase II che gli antiossidanti sono in grado di correggere lo stress ossidativo cronico e anche di far rivivere un normale sistema redox.
GSK3? sotto la gestione di AKT e PI3K, fosforila Fyn con conseguente localizzazione nucleare di Fyn, che Fyn fosforila Nrf2Y568 portando all'esportazione nucleare e alla degradazione di Nrf2.
NRF2 attenua anche la risposta TH1 / TH17 e arricchisce la risposta TH2.
Gli inibitori dell'HDAC hanno attivato la via di segnalazione Nrf2 e hanno sovra-regolato che il Nrf2 a valle bersaglia la subunità catalitica HO-1, NQO1 e glutammato-cisteina-ligasi o GCLC, frenando Keap1 e incoraggiando la dissociazione di Keap1 da Nrf2, traslocazione nucleare Nrf2 e Nrf2 -ARE vincolante.
Nrf2 include un'emivita di circa 20 minuti in condizioni basali.
Diminuire l'IKK? il pool attraverso l'associazione Keap1 riduce I? B? degradazione e potrebbe essere il meccanismo elusivo mediante il quale è dimostrato che l'attivazione di Nrf2 inibisce l'attivazione di NF-B.
Keap1 non deve sempre essere sottoregolato per far funzionare NRF2, come clorofillina, mirtillo, acido ellagico, astaxantina e polifenoli del tè possono incrementare NRF2 e KEAP1 a percentuali 400.
Nrf2 regola negativamente attraverso il termine di desaturasi di stearoil CoA, o SCD e citrato liasi o CL.
Genetica
KEAP1
rs1048290
Allele C - ha mostrato un rischio significativo e un effetto protettivo contro l'epilessia resistente ai farmaci (DRE)
rs11085735 (Sono AC)
associato con il tasso di declino della funzione polmonare nel LHS
MAPT
rs242561
Allele protettivo dell'allele T per i disturbi parkinsoniani - aveva un legame più forte con NRF2 / sMAF ed era associato con i livelli di mRNA MAPT più alti in 3 diverse regioni del cervello, tra cui la corteccia cerebellare (CRBL), la corteccia temporale (TCTX), la materia bianca intralobulare (WHMT)
NFE2L2 (NRF2)
rs10183914 (Sono CT)
Allele T: aumento dei livelli di proteina Nrf2 e ritardo nell'insorgenza del Parkinson di quattro anni
rs16865105 (Sono AC)
Allele C - ha un rischio più elevato di malattia di Parkinson
rs1806649 (Sono CT)
Allele C - è stato identificato e potrebbe essere rilevante per l'eziologia del tumore della mammella.
associato ad un aumentato rischio di ricoveri ospedalieri durante periodi di alti livelli di PM10
rs1962142 (Sono GG)
Allele T - era associato ad un basso livello di espressione citoplasmatica di NRF2 (P = 0.036) ed espressione di solfiredossina negativa (P = 0.042)
Un allele - protetto dal declino del flusso sanguigno dell'avambraccio (FEV) (volume espiratorio forzato in un secondo) in relazione allo stato di tabagismo (p = 0.004)
rs2001350 (Sono TT)
Allele T - protetto dal declino del FEV (volume espiratorio forzato in un secondo) in relazione allo stato di tabagismo (p = 0.004)
rs2364722 (Sono AA)
Un allele - protetto dal declino del FEV (volume espiratorio forzato in un secondo) in relazione allo stato di tabagismo (p = 0.004)
rs2364723
Allele C - associato a FEV significativamente ridotto nei fumatori giapponesi con cancro del polmone
rs2706110
G allele - ha mostrato un rischio significativo e un effetto protettivo contro l'epilessia resistente ai farmaci (DRE)
Alleli AA: hanno mostrato un'espressione KEAP1 significativamente ridotta
AA alleli - era associato ad un aumentato rischio di cancro al seno (P = 0.011)
rs2886161 (Sono TT)
Allele T - associato alla malattia di Parkinson
rs2886162
Un allele - era associato a una bassa espressione di NRF2 (P = 0.011; OR, 1.988; CI, 1.162-3.400) e il genotipo AA era associato a una sopravvivenza peggiore (P = 0.032; HR, 1.687; CI, 1.047-2.748)
rs35652124 (Sono TT)
Un allele - associato a un più alto associato con l'età di esordio per il morbo di Parkinson vs G allele
Allele C - aveva aumentato la proteina NRF2
Allele T: aveva meno proteine NRF2 e un maggior rischio di malattie cardiache e pressione arteriosa
rs6706649 (sono CC)
Allele C - aveva una proteina NRF2 inferiore e aumenta il rischio di malattia di Parkinson
rs6721961 (Sono GG)
Allele T: aveva una proteina NRF2 più bassa
Alleli TT - associazione tra fumo di sigaretta nei forti fumatori e diminuzione della qualità dello sperma
Allele TT - era associato ad un aumento del rischio di cancro al seno [P = 0.008; OR 4.656; intervallo di confidenza (CI), 1.350-16.063] e l'allele T era associato a una bassa estensione dell'espressione della proteina NRF2 (P = 0.0003; OR, 2.420; CI, 1.491-3.926) e all'espressione SRXN1 negativa (P = 0.047; OR, 1.867; CI = 1.002-3.478)
Allele T - allele era anche nominalmente associato a mortalità 28-day associata a ALI in seguito a sindrome da risposta infiammatoria sistemica
Allele T - protetto dal declino del FEV (volume espiratorio forzato in un secondo) in relazione allo stato di tabagismo (p = 0.004)
Allele G - associato ad un aumentato rischio di ALI a seguito di traumi importanti in Europa e afro-americani (odds ratio, OR 6.44; 95% intervallo di confidenza
Alleli AA - associati all'asma indotto da infezione
Alleli AA - mostravano un'espressione del gene NRF2 significativamente ridotta e, di conseguenza, un aumento del rischio di cancro ai polmoni, specialmente quelli che avevano mai fumato
AA alleli - ha avuto un rischio significativamente più elevato per lo sviluppo di T2DM (O 1.77; 95% CI 1.26, 2.49; p = 0.011) rispetto a quelli con il genotipo CC
Alleli AA: forte associazione tra riparazione delle ferite e tossicità tardiva delle radiazioni (associata a un rischio significativamente più elevato di sviluppare effetti tardivi negli afro-americani con una tendenza nei caucasici)
associato alla terapia con estrogeni orali e al rischio di tromboembolia venosa nelle donne in postmenopausa
rs6726395 (Sono AG)
Allele - protetto dal declino di FEV1 (volume espiratorio forzato in un secondo) in relazione allo stato di tabagismo (p = 0.004)
Un allele - associato a FEV1 significativamente ridotto nei fumatori giapponesi con cancro del polmone
GG alleli - avevano livelli più elevati di NRF2 e diminuito rischio di degenerazione maculare
Alleli GG - avevano una maggiore sopravvivenza con colangiocarcinoma
rs7557529 (Sono CT)
Allele C - associato alla malattia di Parkinson
Lo stress ossidativo e altri fattori di stress possono causare danni alle cellule che possono portare a una serie di problemi di salute. Studi di ricerca hanno dimostrato che l'attivazione di Nrf2 può promuovere il meccanismo antiossidante protettivo del corpo umano, tuttavia, i ricercatori hanno discusso sul fatto che la sovraespressione di Nrf2 può avere enormi rischi per la salute e il benessere generale. Vari tipi di cancro possono anche verificarsi con l'overactivation di Nrf2.
Dr. Alex Jimenez DC, CCST Insight
Sulforafano e suoi effetti su cancro, mortalità, invecchiamento, cervello e comportamento, malattie cardiache e altro
Gli isotiocianati sono alcuni dei composti vegetali più importanti che si possono ottenere nella dieta. In questo video faccio il caso più completo per loro che sia mai stato fatto. Soglia di attenzione breve? Passa al tuo argomento preferito facendo clic su uno dei seguenti punti temporali. Timeline completa di seguito.
Sezioni chiave:
00: 01: 14 - Cancro e mortalità
00: 19: 04 - Invecchiamento
00: 26: 30 - Cervello e comportamento
00: 38: 06 - Riassunto finale
00: 40: 27 - Dose
Timeline completa:
00: 00: 34 - Introduzione di sulforaphane, uno degli obiettivi principali del video.
00: 01: 14 - Consumo di verdure crocifere e riduzione della mortalità per tutte le cause.
00: 02: 12 - Rischio di cancro alla prostata.
00: 02: 23 - Rischio di cancro alla vescica.
00: 02: 34 - Carcinoma polmonare nei fumatori.
00: 02: 48 - Rischio di cancro al seno.
00: 03: 13 - Ipotetico: cosa succede se hai già un cancro? (Interventistica)
00: 03: 35 - Meccanismo plausibile che guida i dati associativi sul cancro e sulla mortalità.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Prova animale che mostra un forte effetto dell'estratto di germogli di broccolo sullo sviluppo del tumore della vescica nei ratti.
00: 06: 06 - Effetto dell'integrazione diretta di sulforafano nei pazienti affetti da cancro alla prostata.
00: 07: 09 - Bioaccumulo di metaboliti di isotiocianato nel tessuto mammario effettivo.
00: 08: 32 - Inibizione delle cellule staminali del carcinoma mammario.
00: 08: 53 - Lezione di storia: le brassiche sono state istituite come aventi proprietà sanitarie anche nell'antica Roma.
00: 09: 16 - La capacità del Sulforaphane di potenziare l'escrezione di cancerogeno (benzene, acroleina).
00: 09: 51 - NRF2 come interruttore genetico tramite elementi di risposta antiossidante.
00: 10: 10 - Come l'attivazione di NRF2 migliora l'escrezione di cancerogeno tramite glutatione-S-coniugati.
00: 10: 34 - I cavoletti di Bruxelles aumentano la glutatione-S-transferasi e riducono il danno al DNA.
00: 11: 20 - La bevanda di germogli di broccoli aumenta l'escrezione di benzene di 61%.
00: 13: 31 - L'omogenato di germogli di broccoli aumenta gli enzimi antiossidanti nelle vie aeree superiori.
00: 15: 45 - Consumo di verdure crocifere e mortalità per malattie cardiache.
00: 16: 55 - La polvere di germogli di broccoli migliora i lipidi nel sangue e il rischio complessivo di malattie cardiache nei diabetici di tipo 2.
00: 19: 04 - Inizio della sezione di invecchiamento.
00: 19: 21 - La dieta arricchita con sulforafano migliora la durata della vita dei coleotteri da 15 a 30% (in determinate condizioni).
00: 20: 34 - L'importanza della bassa infiammazione per la longevità.
00: 22: 05 - Le verdure crocifere e la polvere di germogli di broccoli sembrano ridurre un'ampia varietà di marcatori infiammatori negli esseri umani.
00: 23: 40 - Ricapitolazione di metà video: cancro, sezioni di invecchiamento
00: 24: 14 - Gli studi sui topi suggeriscono che il sulforafano potrebbe migliorare la funzione immunitaria adattativa in età avanzata.
00: 25: 18 - Sulforaphane ha migliorato la crescita dei peli in un modello murino di calvizie. Immagine su 00: 26: 10.
00: 26: 30 - Inizio della sezione cervello e comportamento.
00: 27: 18 - Effetto dell'estratto di germogli di broccoli sull'autismo.
00: 27: 48 - Effetto del glucorapanin sulla schizofrenia.
00: 28: 17 - Inizio della discussione sulla depressione (meccanismo e studi plausibili).
00: 31: 21 - Lo studio del mouse utilizzando 10 diversi modelli di depressione indotta da stress mostra sulforapano in modo simile efficace come fluoxetina (prozac).
00: 32: 00 - Lo studio mostra che l'ingestione diretta di glucorafanina nei topi è altrettanto efficace nel prevenire la depressione dal modello di stress sociale di sconfitta.
00: 33: 01 - Inizio della sezione di neurodegenerazione.
00: 33: 30 - Sulforaphane e malattia di Alzheimer.
00: 33: 44 - Sulforaphane e morbo di Parkinson.
00: 33: 51 - Sulforaphane e la malattia di Hungtington.
00: 34: 43 - Inizio della sezione traumatica di lesioni cerebrali.
00: 35: 01 - Sulforaphane iniettato immediatamente dopo TBI migliora la memoria (studio del mouse).
00: 35: 55 - Sulforaphane e plasticità neuronale.
00: 36: 32 - Sulforaphane migliora l'apprendimento nel modello di diabete di tipo II nei topi.
00: 37: 19 - Distrofia muscolare sulforapano e duchenne.
00: 37: 44 - Inibizione della miostatina nelle cellule muscolari satelliti (in vitro).
00: 38: 06 - Ricapitolazione tardiva: mortalità e cancro, danno al DNA, stress ossidativo e infiammazione, escrezione di benzene, malattie cardiovascolari, diabete di tipo II, effetti sul cervello (depressione, autismo, schizofrenia, neurodegenerazione), via NRF2.
00: 40: 27 - Pensieri sulla determinazione di una dose di germogli di broccoli o sulforafano.
00: 41: 01 - Aneddoti su germinazione a casa.
00: 43: 14 - Sulle temperature di cottura e sull'attività del sulforafano.
00: 43: 45 - Conversione batterica intestinale del sulforafano da glucorafanina.
00: 44: 24 - Gli integratori funzionano meglio se combinati con la mirosinasi attiva delle verdure.
00: 44: 56 - Tecniche di cottura e verdure crucifere.
00: 46: 06 - Isotiocianati come goitrogens.
Secondo studi di ricerca, Nrf2, è un fattore di trascrizione fondamentale che attiva i meccanismi antiossidanti protettivi delle cellule per disintossicare il corpo umano. La sovraespressione di Nrf2, tuttavia, può causare problemi di salute. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
A cura di Dr. Alex Jimenez
Discussione aggiuntiva sull'argomento: Dolore alla schiena acuto
Mal di schienaè una delle cause più diffuse di disabilità e di giornate di lavoro perse in tutto il mondo. Il dolore alla schiena si attribuisce al secondo motivo più comune per le visite presso l'ambulatorio medico, superato solo dalle infezioni delle vie respiratorie superiori. Circa l'80% della popolazione sperimenterà dolore alla schiena almeno una volta nella vita. La colonna vertebrale è una struttura complessa composta da ossa, articolazioni, legamenti e muscoli, tra gli altri tessuti molli. Lesioni e / o condizioni aggravate, come dischi erniciati, può eventualmente portare a sintomi di mal di schiena. Le lesioni sportive o gli incidenti automobilistici sono spesso la causa più frequente di mal di schiena, tuttavia a volte il più semplice dei movimenti può avere risultati dolorosi. Fortunatamente, le opzioni di trattamento alternative, come la cura chiropratica, possono aiutare ad alleviare il mal di schiena attraverso l'uso di aggiustamenti spinali e manipolazioni manuali, in definitiva migliorando il sollievo dal dolore.
Molti studi di ricerca attuali sul cancro hanno permesso ai professionisti della salute di capire il modo in cui il corpo detossifica. Analizzando i geni sovraregolati nelle cellule tumorali, i ricercatori hanno scoperto il eritroide nucleare 2 correlato al fattore 2 signaling pathway, meglio noto come Nrf2. NRF2 è un importante fattore di trascrizione che attiva il corpo umano meccanismi antiossidanti protettivi al fine di regolare l'ossidazione da fattori sia esterni che interni per prevenire aumentati livelli di stress ossidativo.
Principi di Nrf2
NRF2 è essenziale per mantenere la salute e il benessere generale perché serve allo scopo principale di regolare il modo in cui gestiamo tutto ciò a cui siamo esposti quotidianamente e non ci ammaliamo. L'attivazione di NRF2 gioca un ruolo nel sistema di disintossicazione di fase II. La disintossicazione di fase II prende i radicali liberi lipofili o liposolubili e li converte in sostanze idrofile o solubili in acqua per l'escrezione, disattivando di conseguenza metaboliti e sostanze chimiche eccezionalmente reattivi della fase I.
L'attivazione di NRF2 riduce l'ossidazione e l'infiammazione complessive del corpo umano attraverso un effetto ormonale. Per innescare NRF2, deve avvenire una reazione infiammatoria dovuta all'ossidazione affinché le cellule producano una risposta adattativa e creino antiossidanti, come il glutatione. Per abbattere il principio di Nrf2, in sostanza, lo stress ossidativo attiva NRF2 che attiva quindi una risposta antiossidante nel corpo umano. NRF2 funziona per bilanciare la segnalazione redox, o l'equilibrio dei livelli di ossidante e antiossidante nella cellula.
Un ottimo esempio di come funziona questo processo può essere dimostrato con l'esercizio. Durante ogni allenamento, il muscolo si adatta in modo da poter accogliere un'altra sessione di allenamento. Se NRF2 diventa sotto o sovraespresso a causa di infezioni croniche o maggiore esposizione alle tossine, che può essere osservato in pazienti con sindrome da risposta infiammatoria cronica, o CIRS, i problemi di salute possono peggiorare a seguito dell'attivazione di NRF2. Soprattutto, se DJ-1 si ossida eccessivamente, l'attivazione di NRF2 terminerà troppo rapidamente.
Effetti dell'attivazione di NRF2
L'attivazione di NRF2 è altamente espressa nei polmoni, nel fegato e nei reni. Eritropatia nucleare Il fattore 2 correlato a 2, o NRF2, funziona più comunemente contrastando l'aumento dei livelli di ossidazione nel corpo umano che può portare allo stress ossidativo. L'attivazione di Nrf2 può aiutare a trattare una varietà di problemi di salute, tuttavia, l'eccessiva attivazione di Nrf2 può peggiorare vari problemi, che sono illustrati di seguito.
L'attivazione periodica di Nrf2 può aiutare:
Invecchiamento (cioè longevità)
Autoimmunità e infiammazione generale (cioè artrite, autismo)
Trapianto di cuore (mentre NRF2 aperto può essere cattivo, NRF2 può aiutare con la riparazione)
Epatite C
Nefrite (casi gravi)
Vitiligine
Inoltre, NRF2 può aiutare a far funzionare integratori nutrizionali, farmaci e farmaci specifici. Molti integratori naturali possono anche aiutare a innescare NRF2. Attraverso gli attuali studi di ricerca, i ricercatori hanno dimostrato che un gran numero di composti che una volta si credeva fossero antiossidanti erano in realtà pro-ossidanti. Questo perché quasi tutti hanno bisogno di NRF2 per funzionare, anche integratori come la curcumina e l'olio di pesce. Il cacao, ad esempio, ha dimostrato di generare effetti antiossidanti nei topi che possiedono il gene NRF2.
Modi per attivare NRF2
Nel caso di malattie neurodegenerative come il morbo di Alzheimer, il morbo di Parkinson, l'ictus o anche le malattie autoimmuni, è probabilmente meglio avere upfoleur Nrf2, ma in modo ormonico. La miscelazione degli attivatori NRF2 può anche avere un effetto additivo o sinergico, poiché occasionalmente può essere dose-dipendente. I modi migliori per aumentare l'espressione di Nrf2 sono elencati di seguito:
HIST (Esercizio) + CoQ10 + Sun (questi si sincronizzano molto bene)
Germogli di broccoli + LLLT sulla mia testa e budello
Butirrato + Super caffè + Morning Sun
L'agopuntura (questo è un metodo alternativo, l'agopuntura laser può anche essere usata)
Semi-Digiuno
cannabidiolo (CBD)
Lion's Mane + Melatonin
Acido alfa-lipoico + DIM
Assenzio romano
Attivazione PPAR-gamma
Il seguente elenco completo contenente su 350 altri modi per attivare Nrf2 attraverso dieta, stile di vita e dispositivi, probiotici, integratori, erbe e oli, ormoni e neurotrasmettitori, farmaci / farmaci e prodotti chimici, fattori di trascrizione / pathway, così come altri modi, è solo una breve guida su cosa può innescare Nrf2. Per brevità in questo articolo, abbiamo omesso su 500 altri alimenti, supplementi nutrizionali e composti che possono aiutare ad attivare Nrf2. Di seguito sono elencati:
Dieta:
Bacche di acai
Alcool (Il vino rosso è migliore, specialmente se vi è un tappo, poiché l'aldeide protocatechuic dai tappi può anche attivare NRF2 .In generale, l'alcol non è raccomandato, sebbene l'assunzione acuta aumenti NRF2.L'assunzione cronica può ridurre NRF2.
Alghe (fuco)
Mele
Tè nero
Noci del Brasile
Germogli di broccoli (e altri isotiocianati, sulforaphane e verdure crocifere come bok choy che hanno D3T)
Mirtilli (0.6-10 g / giorno)
Carote (falcarinone)
Pepe di cayenna (capsaicina)
Sedano (butilftalide)
Chaga (Betulin)
Tè alla camomilla
Chia
Patata cinese
Chokeberries (Aronia)
Cioccolato (scuro o cacao)
Cannella
Caffè (come acido clorogenico, Cafestol e Kahweol)
Cordyceps
Pesce (e frutti di mare)
Di semi di lino
aglio
Ghee (possibilmente)
Ginger (e Cardamonin)
Bacche di Goji
Pompelmo (Naringenin - 50 mg / kg / d naringenin)
Uva
Tè verde
Guaiava
Cuore di palma
Hijiki / Wakame
favo
Kiwi
Legumi
Leone di Leone
Mahuwa
Manghi (Mangiferin)
Mangostano
Latte (capra, mucca - attraverso la regolazione del microbioma)
gelsi
Olio d'oliva (vinacce - idrossitirosolo e acido oleanolico)
Acidi grassi Omega 6 (Lipoxin A4)
Osange Oranges (Morin)
Funghi ostrica
Papaia
Arachidi
Piselli di piccione
Melograno (punicalagina, acido ellagico)
Propoli (Pinocembrin)
Patate dolci viola
Rambutan (Geraniin)
Cipolle
Reishi
Rhodiola Rosea (Salidroside)
Crusca di riso (cycloartenyl ferulate)
Riceberry
Tè Rooibos
Rosmarino
Salvia
cartamo
Olio di sesamo
Soia (e isoflavoni, Daidzein, Genistein)
Squash
Fragole
Tartare di grano saraceno
Timo
Pomodori
Fave di Tonka
Curcuma
Wasabi
Anguria
Stile di vita e dispositivi:
Agopuntura ed elettroagopuntura (tramite collagene a cascata su ECM)
La luce blu
Brain Games (aumenta NRF2 nell'ippocampo)
Restrizione calorica
Freddo (docce, tuffi, bagni di ghiaccio, attrezzi, crioterapie)
EMF (bassa frequenza, come PEMF)
Esercizio (l'esercizio acuto come HIST o HIIT sembra essere più vantaggioso per indurre NRF2, mentre un esercizio più lungo non induce NRF2, ma aumenta i livelli di glutatione)
Dieta ricca di grassi (dieta)
Calore alto (sauna)
Inalazione di idrogeno e acqua di idrogeno
Ossigenoterapia iperbarica
Terapia a infrarossi (come Joovv)
Vitamina C per via endovenosa
dieta chetogenica
Ozono
Fumo (non consigliato - aumento del fumo acuto NRF2, il fumo cronico diminuisce NRF2 Se si sceglie di fumare, Holy Basil può aiutare a proteggere dalla downregulation di NRF2)
Sole (UVB e infrarossi)
Probiotici:
Bacillus subtilis (fmbJ)
Clostridium butyricum (MIYAIRI 588)
Lactobacillus brevis
Lactobacillus casei (SC4 e 114001)
Lactobacillus collinoides
Lactobacillus gasseri (OLL2809, L13-Ia e SBT2055)
Lactobacillus helveticus (NS8)
Lactobacillus paracasei (NTU 101)
Lactobacillus plantarum (C88, CAI6, FC225, SC4)
Lactobacillus rhamnosus (GG)
Integratori, erbe e oli:
Acetil-L-Carnitina (ALCAR) e Carnitina
Allicin
Acido alfa lipoico
amentoflavone
Andrographis paniculata
agmatine
Apigenina
Arginina
Carciofo (Cyanropicrin)
Ashwaganda
Astragalo
Bacopa
Bistecca (Isogemaketone)
Berberine
Beta-cariofillene
Bidens Pilosa
Olio di semi di cumino nero (timoquinone)
Boswellia
Butein
butirrato
cannabidiolo (CBD)
Carotenioidi (come il beta-carotene [sinergia con licopene - 2 15 mg / d licopene], fucoxantina, zeaxantina, astaxantina e luteina)
Chitrak
Chlorella
Clorofilla
Chrysanthemum zawadskii
Cinnamomea
Sundew comune
Rame
coptis
CoQ10
Curcumina
Damiana
Dan Shen / Red Sage (Miltirone)
DIM
Dioscin
Dong Ling Cao
Dong Quai (ginseng femminile)
Ecklonia Cava
EGCG
Elecampane / Inula
Eucommia Bark
Acido ferulico
fisetin
Olio di pesce (DHA / EPA - 3 1 g / d olio di pesce contenente 1098 mg di EPA e 549 mg di DHA)
galangal
Gastrodin (Tian Ma)
Gentiana
Geranio
Ginkgo Biloba (Ginkgolide B)
salicornia
Centella asiatica
Estratto di semi d'uva
Agrimony peloso
Haritaki (Triphala)
Biancospino
Helichrysum
Henna (Juglone)
ibisco
Higenamine
Basilico santo / Tulsi (acido ursolico)
Luppolo
Horny Goat Weed (Icariin / Icariside)
Indigo Naturalis
Ferro (non consigliato se non essenziale)
I3C
Le lacrime di Giobbe
Moringa Oleifera (come Kaempferol)
Inchinkoto (combinazione di Zhi Zi e Wormwood)
Radice di Kudzu
Licorice Root
Radice di Lindera
Luteolina (alte dosi per l'attivazione, basse dosi inibiscono NRF2 nel cancro)
Magnolia
Manjistha
Maximowiczianum (Acerogenina A)
Arnica messicana
Milk Thistle
MitoQ
Mu Xiang
Mucuna pruriens
Nicotinamide e NAD +
Panax Ginseng
Passiflora (come Chrysin, ma il chyrisin può anche ridurre NRF2 attraverso la disregolazione della segnalazione di PI3K / Akt)
Pau d arco (Lapacho)
Phloretin
piceatannolo
PQQ
procyanidin
pterostilbene
Pueraria
Quercetina (solo alte dosi, basse dosi inibiscono NRF2)
Qiang Huo
Red Clover
Resveratrolo (Piceid e altri fitoestrogeni essenzialmente, poligono)
Rosa Canina
Palissandro
Rutina
Sappanwood
Sarsaparilla
Saururus chinensis
SC-E1 (gesso, gelsomino, liquirizia, kudzu e fiori a palloncino)
schisandra
Self Heal (prunella)
Skullcap (Baicalin e Wogonin)
Sheep Sorrel
Si Wu Tang
Sideritis
Spikenard (Aralia)
spirulina
Wort di San Giovanni
sulforafano
Sutherlandia
Tao Hong Si Wu
Taurina
Thunder God Vine (Triptolide)
Tocoferoli (come vitamina E o Linalolo)
Tribulus R
Tu Si Zi
TUDCA
Vitamina A (sebbene altri retinoidi inibiscano NRF2)
Vitamina C (solo dose alta, dose bassa inibisce NRF2)
Vitex / Albero casto
Peonia bianca (Paeoniflorin di Paeonia lactiflora)
Assenzio (Hispidulin e Artemisinin)
Xiao Yao Wan (Wanderer facile e gratuito)
Yerba Santa (Eriodictyol)
Yuan Zhi (Tenuigenin)
Zi Cao (ridurrà NRF2 nel cancro)
Zinco
Ziziphus Jujube
Ormoni e neurotrasmettitori:
L'adiponectina
Adropin
Estrogeno (ma può diminuire NRF2 nel tessuto mammario)
Melatonina
Progesterone
Acido chinolinico (in risposta protettiva per prevenire eccitotossicità)
Serotonina
Gli ormoni tiroidei come T3 (possono aumentare NRF2 in cellule sane, ma diminuirlo nel cancro)
Vitamina D
Farmaci / farmaci e prodotti chimici:
Paracetamolo
Acetazolamide
Amlodipina
auranofin
Bardoxolone metile (BARD)
benznidazole
BHA
CDDO-imidazolide
Ceftriaxone (e antibiotici beta-lattamici)
Cialis
Desametasone
Diprivan (Propofol)
eriodictiolo
Exendin-4
ezetimibe
Fluoruro
fumarato
HNE (ossidato)
Idazoxan
Arsenico inorganico e arsenito di sodio
JQ1 (potrebbe inibire anche NRF2, sconosciuto)
Letairis
melfalan
Methazolamide
Blu di metilene
Nifedipina
FANS
oltipraz
PPI (come Omeprazolo e Lansoprazolo)
Protandim: ottimi risultati in vivo, ma deboli / inesistenti nell'attivazione di NRF2 nell'uomo
Probucol
rapamicina
Reserpina
Rutenio
sitaxentan
Statine (come Lipitor e Simvastatina)
Tamoxifene
Tang Luo Ning
TBHQ
Tecfidera (Dimethyl fumarato)
THC (non forte come CBD)
Teofillina
umbelliferone
Acido Ursodeoxycholic (UDCA)
Verapamil
Viagra
4-Acetoxyphenol
Fattori di trascrizione / percorsi:
? 7 attivazione nAChR
AMPK
La bilirubina
CDK20
CKIP-1
CYP2E1
EAATs
Gankyrin
folletto
GJA1
H-ferritina ferroxidase
Inibitori di HDAC (come acido valproico e TSA, ma possono causare instabilità di NRF2)
Heat Shock Proteins
IL-17
IL-22
Klotho
let-7 (abbatte mBach1 RNA)
MAPK
Michael acceptors (la maggior parte)
miR-141
miR-153
miR-155 (abbatte anche mBach1 RNA)
miR-7 (nel cervello, aiuta con il cancro e la schizofrenia)
Notch1
Stress ossidativo (come ROS, RNS, H2O2) ed elettrofili
PGC-1?
PKC-delta
PPAR-gamma (effetti sinergici)
Inibizione del recettore Sigma-1
SIRT1 (aumenta NRF2 nel cervello e nei polmoni ma può diminuire nel complesso)
SIRT2
SIRT6 (nel fegato e nel cervello)
SRXN1
Inibizione di TrxR1 (attenuazione o esaurimento)
Protoporfirina di zinco
4-HHE
Altro:
Ankaflavin
Amianto
Avicins
Bacillus amyloliquefaciens (usato in agricoltura)
Monossido di carbonio
Daphnetin
Deplezione del glutatione (esaurimento dell'80% 90% possibilmente)
Koraiensis di ginnasta
Epatite C
Herpes (HSV)
Frassino indiano
Indigowoad Root
Isosalipurposide
Isorhamentin
Monascin
Omaveloxolone (forte, alias RTA-408)
PDTC
Carenza di selenio (deficit di selenio può aumentare NRF2)
Larice siberiano
Sophoraflavanone G
Tadehagi triquetrum
Toona sinensis (7-DGD)
Fiore di tromba
63171 e 63179 (forte)
La via di segnalazione 2 del fattore eritroide associato a 2, meglio conosciuta con l'acronimo Nrf2, è un fattore di trascrizione che svolge il ruolo principale di regolazione dei meccanismi antiossidanti protettivi del corpo umano, in particolare per controllare lo stress ossidativo. Mentre l'aumento dei livelli di stress ossidativo può attivare Nrf2, i suoi effetti sono enormemente aumentati attraverso la presenza di composti specifici. Alcuni cibi e integratori aiutano ad attivare Nrf2 nel corpo umano, incluso il isotiocianato sulforafano dai germogli di broccoli. Dr. Alex Jimenez DC, CCST Insight
Sulforafano e suoi effetti su cancro, mortalità, invecchiamento, cervello e comportamento, malattie cardiache e altro
Gli isotiocianati sono alcuni dei composti vegetali più importanti che si possono ottenere nella dieta. In questo video faccio il caso più completo per loro che sia mai stato fatto. Soglia di attenzione breve? Passa al tuo argomento preferito facendo clic su uno dei seguenti punti temporali. Timeline completa di seguito.
Sezioni chiave:
00: 01: 14 - Cancro e mortalità
00: 19: 04 - Invecchiamento
00: 26: 30 - Cervello e comportamento
00: 38: 06 - Riassunto finale
00: 40: 27 - Dose
Timeline completa:
00: 00: 34 - Introduzione di sulforaphane, uno degli obiettivi principali del video.
00: 01: 14 - Consumo di verdure crocifere e riduzione della mortalità per tutte le cause.
00: 02: 12 - Rischio di cancro alla prostata.
00: 02: 23 - Rischio di cancro alla vescica.
00: 02: 34 - Carcinoma polmonare nei fumatori.
00: 02: 48 - Rischio di cancro al seno.
00: 03: 13 - Ipotetico: cosa succede se hai già un cancro? (Interventistica)
00: 03: 35 - Meccanismo plausibile che guida i dati associativi sul cancro e sulla mortalità.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Prova animale che mostra un forte effetto dell'estratto di germogli di broccolo sullo sviluppo del tumore della vescica nei ratti.
00: 06: 06 - Effetto dell'integrazione diretta di sulforafano nei pazienti affetti da cancro alla prostata.
00: 07: 09 - Bioaccumulo di metaboliti di isotiocianato nel tessuto mammario effettivo.
00: 08: 32 - Inibizione delle cellule staminali del carcinoma mammario.
00: 08: 53 - Lezione di storia: le brassiche sono state istituite come aventi proprietà sanitarie anche nell'antica Roma.
00: 09: 16 - La capacità del Sulforaphane di potenziare l'escrezione di cancerogeno (benzene, acroleina).
00: 09: 51 - NRF2 come interruttore genetico tramite elementi di risposta antiossidante.
00: 10: 10 - Come l'attivazione di NRF2 migliora l'escrezione di cancerogeno tramite glutatione-S-coniugati.
00: 10: 34 - I cavoletti di Bruxelles aumentano la glutatione-S-transferasi e riducono il danno al DNA.
00: 11: 20 - La bevanda di germogli di broccoli aumenta l'escrezione di benzene di 61%.
00: 13: 31 - L'omogenato di germogli di broccoli aumenta gli enzimi antiossidanti nelle vie aeree superiori.
00: 15: 45 - Consumo di verdure crocifere e mortalità per malattie cardiache.
00: 16: 55 - La polvere di germogli di broccoli migliora i lipidi nel sangue e il rischio complessivo di malattie cardiache nei diabetici di tipo 2.
00: 19: 04 - Inizio della sezione di invecchiamento.
00: 19: 21 - La dieta arricchita con sulforafano migliora la durata della vita dei coleotteri da 15 a 30% (in determinate condizioni).
00: 20: 34 - L'importanza della bassa infiammazione per la longevità.
00: 22: 05 - Le verdure crocifere e la polvere di germogli di broccoli sembrano ridurre un'ampia varietà di marcatori infiammatori negli esseri umani.
00: 23: 40 - Ricapitolazione di metà video: cancro, sezioni di invecchiamento
00: 24: 14 - Gli studi sui topi suggeriscono che il sulforafano potrebbe migliorare la funzione immunitaria adattativa in età avanzata.
00: 25: 18 - Sulforaphane ha migliorato la crescita dei peli in un modello murino di calvizie. Immagine su 00: 26: 10.
00: 26: 30 - Inizio della sezione cervello e comportamento.
00: 27: 18 - Effetto dell'estratto di germogli di broccoli sull'autismo.
00: 27: 48 - Effetto del glucorapanin sulla schizofrenia.
00: 28: 17 - Inizio della discussione sulla depressione (meccanismo e studi plausibili).
00: 31: 21 - Lo studio del mouse utilizzando 10 diversi modelli di depressione indotta da stress mostra sulforapano in modo simile efficace come fluoxetina (prozac).
00: 32: 00 - Lo studio mostra che l'ingestione diretta di glucorafanina nei topi è altrettanto efficace nel prevenire la depressione dal modello di stress sociale di sconfitta.
00: 33: 01 - Inizio della sezione di neurodegenerazione.
00: 33: 30 - Sulforaphane e malattia di Alzheimer.
00: 33: 44 - Sulforaphane e morbo di Parkinson.
00: 33: 51 - Sulforaphane e la malattia di Hungtington.
00: 34: 43 - Inizio della sezione traumatica di lesioni cerebrali.
00: 35: 01 - Sulforaphane iniettato immediatamente dopo TBI migliora la memoria (studio del mouse).
00: 35: 55 - Sulforaphane e plasticità neuronale.
00: 36: 32 - Sulforaphane migliora l'apprendimento nel modello di diabete di tipo II nei topi.
00: 37: 19 - Distrofia muscolare sulforapano e duchenne.
00: 37: 44 - Inibizione della miostatina nelle cellule muscolari satelliti (in vitro).
00: 38: 06 - Ricapitolazione tardiva: mortalità e cancro, danno al DNA, stress ossidativo e infiammazione, escrezione di benzene, malattie cardiovascolari, diabete di tipo II, effetti sul cervello (depressione, autismo, schizofrenia, neurodegenerazione), via NRF2.
00: 40: 27 - Pensieri sulla determinazione di una dose di germogli di broccoli o sulforafano.
00: 41: 01 - Aneddoti su germinazione a casa.
00: 43: 14 - Sulle temperature di cottura e sull'attività del sulforafano.
00: 43: 45 - Conversione batterica intestinale del sulforafano da glucorafanina.
00: 44: 24 - Gli integratori funzionano meglio se combinati con la mirosinasi attiva delle verdure.
00: 44: 56 - Tecniche di cottura e verdure crucifere.
00: 46: 06 - Isotiocianati come goitrogens.
Secondo molti studi di ricerca attuali, la via di segnalazione del fattore 2 correlata all'eritroide nucleare 2, meglio conosciuta come Nrf2, è un fattore di trascrizione fondamentale che attiva i meccanismi antiossidanti protettivi delle cellule per disintossicare il corpo umano da fattori sia esterni che interni e prevenire l'aumento livelli di stress ossidativo. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
A cura di Dr. Alex Jimenez
Discussione aggiuntiva sull'argomento: Dolore alla schiena acuto
Mal di schienaè una delle cause più diffuse di disabilità e di giornate di lavoro perse in tutto il mondo. Il dolore alla schiena si attribuisce al secondo motivo più comune per le visite presso l'ambulatorio medico, superato solo dalle infezioni delle vie respiratorie superiori. Circa l'80% della popolazione sperimenterà dolore alla schiena almeno una volta nella vita. La colonna vertebrale è una struttura complessa composta da ossa, articolazioni, legamenti e muscoli, tra gli altri tessuti molli. Lesioni e / o condizioni aggravate, come dischi erniciati, può eventualmente portare a sintomi di mal di schiena. Gli infortuni sportivi o gli incidenti automobilistici sono spesso la causa più frequente di mal di schiena, tuttavia, a volte il più semplice dei movimenti può avere risultati dolorosi. Fortunatamente, opzioni di trattamento alternative, come la cura chiropratica, possono aiutare ad alleviare il mal di schiena attraverso l'uso di aggiustamenti spinali e manipolazioni manuali, migliorando in definitiva il sollievo dal dolore.
Lo stress ossidativo è un importante contributo allo sviluppo di una varietà di problemi di salute, tra cui il cancro, le malattie cardiache, il diabete, l'invecchiamento accelerato e la neurodegenerazione. Alimenti ricchi di antiossidanti, erbe e integratori possono essere utilizzati per proteggere il corpo umano da elevati livelli di stress ossidativo. Recenti studi di ricerca hanno dimostrato che il Via del gene Nrf2 può aiutare ad amplificare gli effetti degli antiossidanti. Il Benefici di Nrf2 sono descritti di seguito.
Protegge il corpo contro le tossine
NRF2 è una sostanza intrinseca in grado di proteggere le cellule da composti nocivi, interni ed esterni. NRF2 può aiutare ad arricchire la reazione del corpo umano a farmaci / farmaci e tossine, migliorando la produzione di proteine che aiutano ad eliminare i composti dalla cellula, noti come proteine associate alla resistenza multipla o MRP. Ad esempio, NRF2 viene attivato su inalazione di fumo di sigaretta per consentire ai polmoni di disintossicarsi.
Inoltre, è essenziale che i polmoni si proteggano da allergeni, malattie virali, endotossine batteriche, iperossia e vari inquinanti ambientali. Il fattore scatenante costante di Nrf2, tuttavia, può ridurre i livelli di una sostanza nota come glutatione in tutto il corpo umano. NRF2 può anche proteggere il fegato dalla tossicità e può proteggere il fegato dall'epatotossicità dell'arsenico. Inoltre, NRF2 protegge il fegato e il cervello dal consumo di alcol. Ad esempio, Nrf2 può proteggere dalla tossicità del paracetamolo.
Combatte l'infiammazione e lo stress ossidativo
L'attivazione di NRF2 può aiutare a combattere l'infiammazione diminuendo le citochine infiammatorie, come quelle presenti nella psoriasi. NRF2 può anche ridurre l'infiammazione associata a una varietà di problemi di salute come l'artrite e la fibrosi del fegato, dei reni e dei polmoni. NRF2 può anche aiutare a controllare le allergie abbassando le citochine Th1 / Th17 e aumentando le citochine TH2. Questo può essere utile per disturbi come l'asma.
NRF2 protegge inoltre dai danni cellulari causati dalla luce blu e dai raggi UVA / UVB presenti nella luce solare. Le carenze di Nrf2 possono rendere molto più facile scottarsi. Una delle ragioni alla base di ciò è che NRF2 è in grado di regolare il collagene in risposta alla radiazione UV. I prodotti finali della glicazione avanzata, o AGE, contribuiscono allo sviluppo di molti problemi di salute, tra cui il diabete e le malattie neurodegenerative. NRF2 può ridurre lo stress ossidativo degli AGE all'interno del corpo. NRF2 può anche proteggere il corpo umano da livelli più elevati di stress dovuto al calore.
Migliora i mitocondri e le prestazioni degli esercizi
NRF2 è un potenziatore mitocondriale. L'attivazione di NRF2 contribuisce a un aumento dell'energia ATP per i mitocondri, oltre a un maggiore uso di ossigeno, citrato e grasso. Senza NRF2, i mitocondri avrebbero solo la capacità di funzionare con lo zucchero, o il glucosio, piuttosto che con i grassi. NRF2 è anche essenziale per lo sviluppo dei mitocondri attraverso un processo noto come biogenesi. L'attivazione di NRF2 è vitale per trarre vantaggio dai benefici dell'esercizio.
A causa dell'attività di NRF2, l'esercizio aumenta la funzione mitocondriale, dove questo risultato può essere amplificato con CoQ10, Cordyceps e Caloric Restriction. L'esercizio moderato o l'esercizio acuto inducono la biogenesi mitocondriale e un'elevata sintesi di superossido dismutasi, o SOD, e eme-ossigenasi-1, o HO-1, attraverso l'attivazione di NRF2. L'acido alfa-lipoico, o ALA, e Dan Shen possono potenziare la biogenesi mitocondriale mediata da NRF2. Inoltre, NRF2 può anche migliorare la tolleranza all'esercizio laddove l'eliminazione di NRF2 rende l'esercizio dannoso.
Protegge dall'ipossia
NRF2 aiuta anche a proteggere il corpo umano dalla perdita / esaurimento di ossigeno cellulare, un problema di salute chiamato ipossia. Gli individui con CIRS hanno livelli ridotti di ossigeno poiché il loro NRF2 è ostruito, con conseguente riduzione dei livelli di VEGF, HIF1 e HO-1. Ordinariamente, in individui sani con ipossia, miR-101, che è richiesto per la creazione di cellule staminali, sono sovraespressi e aumentano le quantità di NRF2 / HO-1 e VEGF / eNOS, quindi prevengono danni cerebrali, ma ciò non sembra verificarsi in CIRS.
L'ipossia, caratterizzata da un basso HIF1, nel CIRS può anche causare una perdita di sangue nella barriera emato-encefalica a causa di uno squilibrio NRF2. Il salidroside, situato nella Rhodiola, funziona sull'attivazione di NRF2 e aiuta l'ipossia aumentando i livelli di VEGF e HIF1 all'interno del corpo umano. NRF2 può anche proteggere l'accumulo di lattato nel cuore. L'attivazione di NRF2 può anche interrompere la malattia del movimento di altitudine indotta dall'ipossia, o AMS.
Rallenta l'invecchiamento
Diversi composti che possono essere fatali in quantità massicce possono aumentare la longevità in quantità piuttosto piccole a causa della xenoormesi attraverso NRF2, PPAR-gamma e FOXO. Una quantità molto piccola di tossine aumenta la capacità della cellula di diventare meglio equipaggiata per la prossima volta che viene sfidata con una tossina, tuttavia, questa non è un'approvazione a consumare sostanze chimiche velenose.
Un buon esempio di questo processo è con la restrizione calorica. NRF2 può migliorare la durata della vita delle cellule aumentando i loro livelli di mitocondri e antiossidanti e riducendo la capacità delle cellule di morire. NRF2 diminuisce con l'invecchiamento perché NRF2 previene la morte delle cellule staminali e le aiuta a rigenerarsi. NRF2 svolge un ruolo nel migliorare la guarigione delle ferite.
Aumenta il sistema vascolare
Effettuato correttamente con la produzione di sulforafano, l'attivazione di NRF2 può proteggere da malattie cardiache quali ipertensione, ipertensione e indurimento delle arterie o aterosclerosi. NRF2 può migliorare l'attività dell'acetilcolina, o ACh, rilassante sul sistema vascolare riducendo lo stress indotto dal colesterolo. L'attivazione di Nrf2 può rafforzare il cuore, tuttavia, Nrf2 sovraattivato può aumentare la probabilità di malattie cardiovascolari.
Le statine possono prevenire o portare a malattie cardiovascolari. NRF2 svolge anche un ruolo importante nel bilanciare ferro e calcio che possono proteggere il corpo umano da livelli elevati di ferro. Ad esempio, Sirtuin 2, o SIRT2, è in grado di regolare l'omeostasi del ferro nelle cellule mediante l'attivazione di NRF2 che si ritiene sia necessaria per livelli sani di ferro. NRF2 può anche aiutare con Sickle Cell Disease, o SCD. La disfunzione di NRF2 potrebbe essere un motivo alla base dell'endotossemia come la disbiosi o l'ipertensione indotta da lectine. Nrf2 può anche proteggere il corpo umano dai danni indotti dall'anfetamina al sistema vascolare.
Combatte la neuroinfiammazione
NRF2 può schermare e aiutare con l'infiammazione del cervello, comunemente indicata come neuroinfiammazione. Inoltre, NRF2 può aiutare con un Assortimento di Sistema Nervoso Centrale, o CNS, disturbi, tra cui:
La malattia di Alzheimer (AD) - riduce lo stress beta-amiloide sui mitocondri
Sclerosi Laterale Amiotrofica (SLA)
La malattia di Huntington (HD)
La sclerosi multipla (SM)
Nerve Regeneration
Morbo di Parkinson (PD) - protegge la dopamina
Infortunio al midollo spinale (SCI)
Ictus (ischemico ed emorragico) - aiuta l'ipossia
Brain Injury traumatica
NRF2 ha rivelato una diminuzione della neuroinfiammazione negli adolescenti con disturbi dello spettro autistico o ASD. L'idebenone si accoppia correttamente con gli attivatori NRF2 contrari alla neuroinfiammazione. NRF2 può anche migliorare la barriera emato-encefalica, o BBB. Ad esempio, l'attivazione di NRF2 con acido carnosico ottenuto da rosmarino e salvia può attraversare il BBB e causare neurogenesi. È stato anche dimostrato che NRF2 aumenta il fattore neurotrofico derivato dal cervello, o BDNF.
NRF2 modula anche la capacità di alcuni integratori nutrizionali di causare il fattore di crescita nervoso, o NGF, poiché può anche aiutare con la nebbia cerebrale e problemi indotti dal glutammato modulando i recettori N-metil-D-aspartato o NMDA. Può anche ridurre lo stress ossidativo da acido chinolinico, denominato QUIN. L'attivazione di NRF2 può proteggere dalle convulsioni e dosi elevate possono ridurre l'orlo di una crisi. A dosi regolari di stimolazione, NRF2 può migliorare le capacità cognitive a seguito di una crisi convulsiva abbassando il glutammato extracellulare nel cervello e grazie alla sua capacità di trarre la cisteina dal glutammato e dal glutatione.
Allevia la depressione
Nella depressione, è normale notare un'infiammazione nel cervello, specialmente dalla corteccia prefrontale e dall'ippocampo, così come una riduzione del BDNF. In alcune versioni della depressione, NRF2 può migliorare i sintomi depressivi abbassando l'infiammazione all'interno del cervello e aumentando i livelli di BDNF. La capacità di agmatina di ridurre la depressione aumentando la noradrenalina, la dopamina, la serotonina e il BDNF nell'ippocampo dipende dall'attivazione di NRF2.
Contiene proprietà anti-cancro
NRF2 è ugualmente un soppressore del tumore in quanto è un promotore del tumore se non gestito di conseguenza. NRF2 può proteggere contro il cancro causato dai radicali liberi e dallo stress ossidativo, tuttavia, la sovraespressione di NRF2 può essere riscontrata anche nelle cellule tumorali. L'intensa attivazione di NRF2 può aiutare con una varietà di tumori. Ad esempio, il supplemento Protandim può ridurre il cancro della pelle mediante l'attivazione di NRF2.
Allevia il dolore
La malattia della guerra del Golfo, o GWI, una malattia notevole che colpisce i veterani della Guerra del Golfo, è una raccolta di sintomi cronici inspiegabili che possono includere stanchezza, mal di testa, dolori articolari, indigestione, insonnia, vertigini, disturbi respiratori e problemi di memoria. NRF2 può migliorare i sintomi del GWI diminuendo l'infiammazione ippocampale e generale, oltre a diminuire il dolore. NRF2 può inoltre aiutare con il dolore delle lesioni del nervo corporeo e migliorare i danni ai nervi della neuropatia diabetica.
Migliora il diabete
Livelli elevati di glucosio, meglio denominati iperglicemia, provocano danni ossidativi alle cellule a causa dell'interruzione della funzione mitocondriale. L'attivazione di NRF2 può proteggere il corpo umano dai danni dell'iperglicemia alla cellula, prevenendo così la morte cellulare. L'attivazione di NRF2 può inoltre proteggere, ripristinare e migliorare la funzionalità delle cellule beta del pancreas, riducendo al contempo la resistenza all'insulina.
Protegge la vista e l'udito
NRF2 può proteggere dai danni agli occhi causati dalla retinopatia diabetica. Potrebbe anche evitare la formazione di cataratta e proteggere i fotorecettori contrariamente alla morte indotta dalla luce. NRF2 protegge inoltre l'orecchio o la coclea dallo stress e dalla perdita dell'udito.
Potrebbe aiutare l'obesità
NRF2 può aiutare con l'obesità principalmente a causa della sua capacità di regolare le variabili che operano sull'accumulo di grasso nel corpo umano. L'attivazione di NRF2 con sulforafano può aumentare l'inibizione della sintesi degli acidi grassi, o FAS, e le proteine di disaccoppiamento, o UCP, con conseguente minore accumulo di grasso e più grasso bruno, caratterizzato da grasso che include più mitocondri.
Protegge l'intestino
NRF2 aiuta a proteggere l'intestino salvaguardando l'omeostasi del microbioma intestinale. A titolo di esempio, i probiotici lactobacillus attiveranno NRF2 per proteggere l'intestino dallo stress ossidativo. NRF2 può anche aiutare a prevenire la colite ulcerosa o UC.
Protegge gli organi sessuali
NRF2 può proteggere i testicoli e mantenere il numero di spermatozoi da danni nelle persone con diabete. Può anche aiutare con la disfunzione erettile o ED. Alcuni integratori che stimolano la libido come Mucuna, Tribulus e Ashwaganda possono migliorare la funzione sessuale tramite l'attivazione di NRF2. Altri fattori che aumentano NRF2, come la luce solare oi germogli di broccoli, possono anche aiutare a migliorare la libido.
Regola le ossa e i muscoli
Lo stress ossidativo può determinare la densità ossea e la riduzione della forza, che è normale nell'osteoporosi. L'attivazione di NRF2 potrebbe avere la capacità di migliorare gli antiossidanti nelle ossa e proteggere dall'invecchiamento osseo. NRF2 può anche prevenire la perdita muscolare e migliorare la distrofia muscolare di Duchenne, o DMD.
Contiene proprietà anti-virali
Ultimo ma non meno importante, l'attivazione di NRF2 può aiutare a difendere il corpo umano da diversi virus. Nei pazienti con il virus della dengue, i sintomi non erano così intensi negli individui che avevano livelli maggiori di NRF2 rispetto agli individui che avevano meno gradi di NRF2. NRF2 può anche aiutare le persone che hanno il virus dell'immunodeficienza umana-1 o l'HIV. NRF2 può proteggere dallo stress ossidativo del virus adeno-associato, o AAV e H. Pylori. Infine, Lindera Root può sopprimere il virus dell'epatite C con l'attivazione di NRF2.
Nrf2, o fattore 2 correlato a NF-E2, è un fattore di trascrizione trovato nell'uomo che regola l'espressione di un insieme specifico di geni antiossidanti e disintossicanti. Questa via di segnalazione è attivata a causa dello stress ossidativo poiché potenzia numerosi enzimi antiossidanti e di disintossicazione epatica di fase II per ripristinare l'omeostasi nel corpo umano. Gli esseri umani sono adattati per funzionare in uno stato di omeostasi o equilibrio. Quando il corpo si confronta con lo stress ossidativo, Nrf2 si attiva per regolare l'ossidazione e controllare lo stress che provoca. Nrf2 è essenziale per prevenire problemi di salute associati allo stress ossidativo. Dr. Alex Jimenez DC, CCST Insight
Sulforafano e suoi effetti su cancro, mortalità, invecchiamento, cervello e comportamento, malattie cardiache e altro
Gli isotiocianati sono alcuni dei composti vegetali più importanti che si possono ottenere nella dieta. In questo video faccio il caso più completo per loro che sia mai stato fatto. Soglia di attenzione breve? Passa al tuo argomento preferito facendo clic su uno dei seguenti punti temporali. Timeline completa di seguito.
Sezioni chiave:
00: 01: 14 - Cancro e mortalità
00: 19: 04 - Invecchiamento
00: 26: 30 - Cervello e comportamento
00: 38: 06 - Riassunto finale
00: 40: 27 - Dose
Timeline completa:
00: 00: 34 - Introduzione di sulforaphane, uno degli obiettivi principali del video.
00: 01: 14 - Consumo di verdure crocifere e riduzione della mortalità per tutte le cause.
00: 02: 12 - Rischio di cancro alla prostata.
00: 02: 23 - Rischio di cancro alla vescica.
00: 02: 34 - Carcinoma polmonare nei fumatori.
00: 02: 48 - Rischio di cancro al seno.
00: 03: 13 - Ipotetico: cosa succede se hai già un cancro? (Interventistica)
00: 03: 35 - Meccanismo plausibile che guida i dati associativi sul cancro e sulla mortalità.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Prova animale che mostra un forte effetto dell'estratto di germogli di broccolo sullo sviluppo del tumore della vescica nei ratti.
00: 06: 06 - Effetto dell'integrazione diretta di sulforafano nei pazienti affetti da cancro alla prostata.
00: 07: 09 - Bioaccumulo di metaboliti di isotiocianato nel tessuto mammario effettivo.
00: 08: 32 - Inibizione delle cellule staminali del carcinoma mammario.
00: 08: 53 - Lezione di storia: le brassiche sono state istituite come aventi proprietà sanitarie anche nell'antica Roma.
00: 09: 16 - La capacità del Sulforaphane di potenziare l'escrezione di cancerogeno (benzene, acroleina).
00: 09: 51 - NRF2 come interruttore genetico tramite elementi di risposta antiossidante.
00: 10: 10 - Come l'attivazione di NRF2 migliora l'escrezione di cancerogeno tramite glutatione-S-coniugati.
00: 10: 34 - I cavoletti di Bruxelles aumentano la glutatione-S-transferasi e riducono il danno al DNA.
00: 11: 20 - La bevanda di germogli di broccoli aumenta l'escrezione di benzene di 61%.
00: 13: 31 - L'omogenato di germogli di broccoli aumenta gli enzimi antiossidanti nelle vie aeree superiori.
00: 15: 45 - Consumo di verdure crocifere e mortalità per malattie cardiache.
00: 16: 55 - La polvere di germogli di broccoli migliora i lipidi nel sangue e il rischio complessivo di malattie cardiache nei diabetici di tipo 2.
00: 19: 04 - Inizio della sezione di invecchiamento.
00: 19: 21 - La dieta arricchita con sulforafano migliora la durata della vita dei coleotteri da 15 a 30% (in determinate condizioni).
00: 20: 34 - L'importanza della bassa infiammazione per la longevità.
00: 22: 05 - Le verdure crocifere e la polvere di germogli di broccoli sembrano ridurre un'ampia varietà di marcatori infiammatori negli esseri umani.
00: 23: 40 - Ricapitolazione di metà video: cancro, sezioni di invecchiamento
00: 24: 14 - Gli studi sui topi suggeriscono che il sulforafano potrebbe migliorare la funzione immunitaria adattativa in età avanzata.
00: 25: 18 - Sulforaphane ha migliorato la crescita dei peli in un modello murino di calvizie. Immagine su 00: 26: 10.
00: 26: 30 - Inizio della sezione cervello e comportamento.
00: 27: 18 - Effetto dell'estratto di germogli di broccoli sull'autismo.
00: 27: 48 - Effetto del glucorapanin sulla schizofrenia.
00: 28: 17 - Inizio della discussione sulla depressione (meccanismo e studi plausibili).
00: 31: 21 - Lo studio del mouse utilizzando 10 diversi modelli di depressione indotta da stress mostra sulforapano in modo simile efficace come fluoxetina (prozac).
00: 32: 00 - Lo studio mostra che l'ingestione diretta di glucorafanina nei topi è altrettanto efficace nel prevenire la depressione dal modello di stress sociale di sconfitta.
00: 33: 01 - Inizio della sezione di neurodegenerazione.
00: 33: 30 - Sulforaphane e malattia di Alzheimer.
00: 33: 44 - Sulforaphane e morbo di Parkinson.
00: 33: 51 - Sulforaphane e la malattia di Hungtington.
00: 34: 43 - Inizio della sezione traumatica di lesioni cerebrali.
00: 35: 01 - Sulforaphane iniettato immediatamente dopo TBI migliora la memoria (studio del mouse).
00: 35: 55 - Sulforaphane e plasticità neuronale.
00: 36: 32 - Sulforaphane migliora l'apprendimento nel modello di diabete di tipo II nei topi.
00: 37: 19 - Distrofia muscolare sulforapano e duchenne.
00: 37: 44 - Inibizione della miostatina nelle cellule muscolari satelliti (in vitro).
00: 38: 06 - Ricapitolazione tardiva: mortalità e cancro, danno al DNA, stress ossidativo e infiammazione, escrezione di benzene, malattie cardiovascolari, diabete di tipo II, effetti sul cervello (depressione, autismo, schizofrenia, neurodegenerazione), via NRF2.
00: 40: 27 - Pensieri sulla determinazione di una dose di germogli di broccoli o sulforafano.
00: 41: 01 - Aneddoti su germinazione a casa.
00: 43: 14 - Sulle temperature di cottura e sull'attività del sulforafano.
00: 43: 45 - Conversione batterica intestinale del sulforafano da glucorafanina.
00: 44: 24 - Gli integratori funzionano meglio se combinati con la mirosinasi attiva delle verdure.
00: 44: 56 - Tecniche di cottura e verdure crucifere.
00: 46: 06 - Isotiocianati come goitrogens.
Quando il corpo umano deve confrontarsi con fattori interni ed esterni dannosi come le tossine, le cellule devono attivare rapidamente le loro capacità antiossidanti per contrastare lo stress ossidativo. Poiché è stato determinato che l'aumento dei livelli di stress ossidativo causa una varietà di problemi di salute, è importante utilizzare l'attivazione Nrf2 per trarne vantaggio. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
A cura di Dr. Alex Jimenez
Discussione aggiuntiva sull'argomento: Dolore alla schiena acuto
Mal di schienaè una delle cause più diffuse di disabilità e di giornate di lavoro perse in tutto il mondo. Il dolore alla schiena si attribuisce alla seconda ragione più comune per le visite mediche, superata solo dalle infezioni delle alte vie respiratorie. Circa l'80% della popolazione sperimenterà dolore alla schiena almeno una volta nella vita. La colonna vertebrale è una struttura complessa composta da ossa, articolazioni, legamenti e muscoli, tra gli altri tessuti molli. Per questo motivo, lesioni e / o condizioni aggravate, come dischi erniciati, può eventualmente portare a sintomi di mal di schiena. Gli infortuni sportivi o gli incidenti automobilistici sono spesso la causa più frequente di mal di schiena, tuttavia, a volte il più semplice dei movimenti può avere risultati dolorosi. Fortunatamente, opzioni di trattamento alternative, come la cura chiropratica, possono aiutare ad alleviare il mal di schiena attraverso l'uso di aggiustamenti spinali e manipolazioni manuali, migliorando in definitiva il sollievo dal dolore.
sulforafano è un phytochemical, una sostanza all'interno del gruppo di isotiocianato di composti organosulfur, trovato in verdure crocifere, come broccoli, cavoli, cavolfiori e cavolini di Bruxelles. Può anche essere trovato in bok choy, kale, cavoli, senape e crescione. Studi di ricerca hanno dimostrato che il sulforafano può aiutare a prevenire vari tipi di cancro entro il 2010 attivando la produzione di Nrf2, o fattore correlato al fattore nucleare eritroide 2, un fattore di trascrizione che regola i meccanismi antiossidanti protettivi che controllano la risposta della cellula agli ossidanti. Lo scopo del seguente articolo è descrivere la funzione del sulforafano.
Astratto
Il sistema antiossidante KEAP1-Nrf2-ARE è un mezzo principale con cui le cellule rispondono agli stress ossidativi e xenobiotici. Il sulforafano (SFN), un isotiocianato elettrofilo derivato da verdure crocifere, attiva la via KEAP1-Nrf2-ARE ed è diventato una molecola di interesse nel trattamento di malattie in cui lo stress ossidativo cronico svolge un ruolo etiologico importante. Dimostriamo qui che i mitocondri di cellule epiteliali della retina umana (RPE-1) in coltura trattate con SFN subiscono l'iperfusione che è indipendente sia da Nrf2 che dal suo inibitore citoplasmatico KEAP1. È stato riportato che la fusione mitocondriale è citoprotettiva inibendo la formazione dei pori nei mitocondri durante l'apoptosi e, coerentemente con questo, mostriamo la citoprotezione indipendente da Nrf2, delle cellule trattate con SFN esposte all'induatore-apoptosi, staurosporina. Meccanicamente, l'SFN attenua il reclutamento e / o la ritenzione del fattore di fissione solubile Drp1 nei mitocondri e nei perossisomi, ma non influenza l'abbondanza complessiva di Drp1. Questi dati dimostrano che le proprietà benefiche di SFN si estendono oltre l'attivazione del sistema KEAP1-Nrf2-ARE e garantiscono ulteriori interrogazioni dato l'uso corrente di questo agente in più studi clinici.
parole chiave:Sulforaphane, Nrf2, Drp1, Mitocondri, Fissione, Fusione, Apoptosi
Introduzione
Sulforaphane è un inibitore indipendente di Nrf2 della fissione mitocondriale
Il sulforafano (SFN) è un composto di isotiocianato derivato dalla dieta più comunemente dalle verdure crocifere [56]. È generato nelle piante come risposta xenobiotica alla predazione tramite rilascio vescicolare dell'enzima idrolitico mirosinasi dalle cellule danneggiate; questo enzima converte i glucosinolati in isotiocianati [42]. Negli ultimi due decenni, SFN è stato ampiamente caratterizzato per le sue proprietà antitumorali, antiossidanti e antimicrobiche segnalate [57]. Gran parte di questa efficacia è stata attribuita alla capacità di SFN di modulare la via di segnalazione dell'elemento di risposta antiossidante (ARE) di KEAP1-Nrf2, sebbene siano state identificate attività aggiuntive del composto, inclusa l'inibizione dell'attività dell'istone deacetilasi e la progressione del ciclo cellulare [ 29]. Nrf2 è il principale fattore di trascrizione antiossidante e in condizioni di omeostasi, la sua stabilità è soppressa attraverso l'azione del complesso citoplasmatico Cullin3KEAP1 ubiquitin ligasi [20]. Nello specifico, Nrf2 viene assunto nella ligasi Cullin3KEAP1 legandosi all'adattatore di substrato dimerico KEAP1 e successivamente modificato con catene polyUb che mirano al fattore di trascrizione per la degradazione mediata dal proteasoma. Questo ricambio costitutivo limita l'emivita di Nrf2 nelle celle non accentate a ~ 15 min [30], [33], [46], [55]. In risposta a numerosi tipi di stress, in particolare lo stress ossidativo, KEAP1, una proteina ricca di cisteina, agisce come un sensore redox e la modificazione ossidativa delle cisteine critiche, in particolare C151, di KEAP1 dissocia Nrf2-KEAP1 da CUL3 prevenendo così la degradazione di Nrf2 [ 8], [20], [55]. In particolare, SFN, e possibilmente altri attivatori Nrf2, imitano lo stress ossidativo modificando C151 di KEAP1 ad esempio [21]. La stabilizzazione di Nrf2 consente la sua traslocazione nel nucleo in cui induce l'espressione di una batteria di geni antiossidanti di fase II e di disintossicazione. Nrf2 si lega agli elementi promotori della risposta antiossidante (ARE) dei suoi geni bersaglio affini attraverso l'eterodimerizzazione con piccole proteine Maf [19]. Questo sistema presenta una risposta dinamica e sensibile agli antiossidanti indiretti come l'SFN, i radicali liberi generati dai mitocondri [16], o altre fonti fisiologiche di stress ossidativo [41].
I mitocondri sono organelli subcellulari dinamici che regolano una serie di funzioni cellulari che vanno dalla produzione di ATP e tampone del calcio intracellulare alla regolazione redox e all'apoptosi [13], [49]. I mitocondri rappresentano anche la principale fonte di specie reattive dell'ossigeno (ROS) all'interno della cellula. È quindi necessaria una corretta regolazione della funzione mitocondriale per ottimizzare la produzione di ATP per soddisfare le esigenze cellulari riducendo al contempo al contempo gli effetti potenzialmente dannosi dell'eccessiva produzione di radicali liberi. Un requisito critico per la modulazione fine della funzione mitocondriale è la capacità dei mitocondri di funzionare sia come macchine biochimiche sia come parte di una vasta rete reattiva.
La morfologia e la funzione della rete mitocondriale sono determinate da un equilibrio regolato tra fissione e fusione. La fissione mitocondriale è necessaria per l'ereditarietà cellulare dei mitocondri durante la divisione cellulare [28] e per la degradazione autofagica selettiva di mitocondri depolarizzati o danneggiati, denominati mitophagy [1]. Al contrario, è necessaria la fusione per la complementazione dei genomi mitocondriali e la condivisione delle componenti della catena di trasporto degli elettroni tra i mitocondri vicini [54]. A livello molecolare, la fissione mitocondriale e la fusione sono regolate da grandi GTPasi di tipo dynaminico. Tre enzimi regolano principalmente la fusione: Mitofusins 1 e 2 (Mfn1 / 2) sono proteine di membrana esterna a due passaggi che mediano la fusione della membrana esterna tramite interazioni eterotipiche tra i mitocondri adiacenti [15], [25], [37], mentre OPA1 è un interno proteina di membrana che assicura simultaneamente la connettività della matrice regolando la fusione delle membrane interne [5]. L'attività GTPase di tutte e tre le proteine è necessaria per la fusione robusta [5], [18] e OPA1 è ulteriormente regolato dalla proteolisi complessa all'interno della membrana mitocondriale da parte delle proteasi OMA1 [14], PARL [6] e YME1L [45 ]. È importante sottolineare che il potenziale di membrana mitocondriale intatto è necessario per una fusione efficiente al fine di sopprimere l'integrazione di mitocondri danneggiati e sani [26].
La fissione mitocondriale è principalmente catalizzata da una proteina citosolica chiamata proteina 1 correlata alla Dynamin (Drp1 / DNM1L). Drp1 è reclutato dal citosol a siti prospettici di fissione sulla membrana esterna mitocondriale [43]. I principali recettori per Drp1 sulla membrana esterna sono il fattore di fissione mitocondriale (Mff) [32] e, in misura minore, Fission 1 (Fis1) [51]. Inoltre, è stato scoperto un recettore dell'esca, MIEF1 / MiD51, che agisce per limitare ulteriormente l'attività della proteina Drp1 in potenziali siti di fissione [58]. Una volta ancorata alla membrana mitocondriale esterna, Drp1 si oligomerizza in strutture a spirale attorno al corpo del mitocondrio e quindi utilizza l'energia derivata dall'idrolisi GTP per mediare la scissione fisica delle membrane mitocondriali esterne e interne [17]. I tubuli derivati dal reticolo endoplasmatico agiscono come un costrittore iniziale dei mitocondri prima dell'oligomerizzazione di Drp1, sottolineando la rivelazione che i mitocondri non ristretti sono più ampi della circonferenza permissiva di una spirale di Drp1 completata [12]. La dinamica dell'actina è anche importante per le interazioni ER-mitocondrio che precedono la fissione mitocondriale [24]. Oltre al suo ruolo nella fissione mitocondriale, Drp1 catalizza la fissione dei perossisomi [40].
Drp1 è molto simile alla ben definita proteina dynamin in quanto entrambe le proteine contengono un dominio GTPase N-terminale, un dominio centrale che è critico per l'auto-oligomerizzazione e un dominio effector GTPase C-terminale [31]. Drp1 raggiunge la selettività per le membrane mitocondriali attraverso una combinazione di interazioni con le sue proteine recettoriali Mff e Fis1 e anche attraverso la sua affinità per la cardiolipina fosfolipidica specifica dei mitocondri attraverso l'esclusivo dominio B-insert di Drp1 [2]. Drp1 esiste tipicamente come un omotetramero nel citoplasma e l'ordine superiore nei siti di fissione mitocondriale è mediato dal dominio centrale di Drp1 [3].
Dato il legame implicito tra la funzione mitocondriale e il percorso di KEAP1-Nrf2-ARE, abbiamo studiato gli effetti dell'attivazione di Nrf2 sulla struttura e la funzione mitocondriale. Dimostriamo qui che SFN induce l'iperfusione mitocondriale che, inaspettatamente, è indipendente da Nrf2 e KEAP1. Questo effetto di SFN avviene attraverso un'inibizione della funzione Drp1. Dimostriamo inoltre che SFN conferisce resistenza all'apoptosi che è Nrf2-indipendente e imita quello osservato nelle cellule impoverito di Drp1. Questi dati indicano collettivamente che oltre a stabilizzare e attivare Nrf2, SFN modula la dinamica mitocondriale e preserva il fitness e la sopravvivenza cellulare.
Risultati
Sulforaphane induce Nrf2 / Iperfusione indipendente di KEAP1 dei mitocondri
Nel corso dello studio degli effetti dell'attivazione di Nrf2 sulle dinamiche della rete mitocondriale, abbiamo scoperto che il trattamento delle cellule epiteliali del pigmento retinico umano immortalate (RPE-1) con sulforafano (SFN), un potente attivatore della segnalazione Nrf2, ha indotto una robusta fusione di la rete mitocondriale rispetto alle cellule di controllo trattate con veicolo (Fig. 1A e B). La morfologia dei mitocondri in queste cellule somigliava molto a quella dei mitocondri nelle cellule impoverite da siRNA di Drp1 endogeno, il principale fattore di fissione mitocondriale (Fig. 1A). Questo risultato ha sollevato l'idea intrigante che la fissione mitocondriale e lo stato di fusione rispondano direttamente ai livelli di Nrf2 nella cellula. Tuttavia, la stimolazione delle cellule con altri stabilizzatori e attivatori Nrf2 come l'inibitore del proteasoma MG132, il pro-ossidante tBHQ o il knockdown dell'inibitore Nrf2 KEAP1 non ha indotto la fusione mitocondriale (Fig. 1A e B). La stabilizzazione di Nrf2 da queste manipolazioni è stata confermata dal western blotting per Nrf2 endogeno (Fig. 1C). Inoltre, l'espressione di Nrf2 era superflua per la fusione mitocondriale indotta da SFN, poiché l'abbattimento di Nrf2 endogeno con siRNA non è riuscito a contrastare questo fenotipo (Fig. 1D F). Poiché SFN stimola la via KEAP1-Nrf2-ARE modificando covalentemente i residui di cisteina di KEAP1 [21], abbiamo abbattuto KEAP1 per stabilire se l'iperfusione mitocondriale indotta da SFN è stimolata attraverso un percorso dipendente da KEAP1, ma Nrf2 indipendente. Tuttavia, anche l'esaurimento di KEAP1 non è riuscito ad abrogare la fusione mitocondriale indotta da SFN (Fig. 1G I). Infatti, SFN ha invertito la morfologia della pro-fissione indotta dall'esaurimento di KEAP1 (Fig. 1G, pannello b contro pannello d). Questi risultati indicano che il trattamento SFN causa la fusione mitocondriale indipendente dal percorso canonico KEAP1-Nrf2-ARE e ci ha portato a interrogare se SFN influenza direttamente i componenti della fissione mitocondriale o del macchinario di fusione.
Figura 1 SFN induce la fusione mitocondriale indipendente da Nrf2 / KEAP1. (A) Le cellule RPE-1 sono state trasfettate con gli siRNA indicati e 3 giorni dopo sono state trattate con DMSO o gli attivatori Nrf2 SFN (50? M), MG132 (10? M) o tBHQ (100? M) per 4 ore. I mitocondri (rossi) sono etichettati con un anticorpo anti-Tom20 e i nuclei (blu) sono controcolorati con DAPI. (B) Grafico che mostra la quantificazione del punteggio morfologico mitocondriale da (A). > 50 cellule per condizione sono state valutate in cieco. (C) Western blot rappresentativi da (A). (D) Le cellule RPE-1 sono state trasfettate con siRNA 10 nM e 3 giorni dopo sono state trattate con SFN per 4 ore prima di essere fissate e colorate come in (A). (E) Grafico che mostra la quantificazione del punteggio del fenotipo mitocondriale da (D). > 100 cellule per condizione sono state valutate in cieco. (F) Western blot rappresentativi da (D). (G) Le cellule sono state trasfettate e trattate come in (D) con siCON o siKEAP1. (H) Le cellule da (G) sono state valutate come in (B) ed (E) sulla base della morfologia mitocondriale. (I) Western blot rappresentativi da (G). I dati in (B), (E) e (H) sono stati compilati da 3 esperimenti indipendenti ciascuno e la significatività statistica è stata determinata dal test t di Student a due code. Le barre di errore riflettono +/- SD (per l'interpretazione dei riferimenti al colore nella legenda di questa figura, si rimanda il lettore alla versione web di questo articolo).
Sulforaphane danneggia l'associazione mitocondriale di Drp1
Sulla base del riscontro che il trattamento con SFN induce l'iperfusione mitocondriale, abbiamo ipotizzato che questo fenotipo fosse o una conseguenza dell'eccessiva attività di fusione o di un'inibizione dell'attività della fissione. Per discriminare tra queste due possibilità, abbiamo confrontato la morfologia dei perossisomi in presenza e assenza di SFN. I perossisomi sono simili ai mitocondri in quanto sono organelli dinamici la cui forma e lunghezza sono costantemente in flusso [44]. I perossisomi contengono sia Fis1 che Mff nella loro membrana esterna e, di conseguenza, sono bersagli per la fissione mediata da Drp1 [22], [23]. Tuttavia, i perossisomi non utilizzano il meccanismo di fusione della rete mitocondriale e, di conseguenza, non subiscono la fusione [39]. Piuttosto, la fissione perossisomale è contrastata dall'allungamento dei perossisomi esistenti attraverso l'aggiunta de novo di membrane e proteine [44]. Poiché ai perossisomi manca Mfn1 / 2 e OPA1, abbiamo pensato che se SFN attivasse il meccanismo di fusione invece di inibire il meccanismo di fissione, la lunghezza del perossisoma non ne risentirebbe. Nelle cellule trattate con veicoli, i perossisomi vengono mantenuti come organuli brevi, rotondi e puntiformi (Fig. 2, pannelli b e d). Tuttavia, il trattamento con SFN ha aumentato la lunghezza del perossisoma di ~ 2-fold rispetto alle cellule di controllo (Fig. 2, pannelli f e h). Inoltre, molti dei perossisomi sono stati pizzicati vicino al centro, indicando un potenziale difetto di scissione (Fig. 2, pannello h, punte di freccia). Allo stesso modo, i perossisomi nelle cellule trasfettate con DrP1 siRNA erano anormalmente lunghi (Fig. 2, pannelli j e l), confermando che Drp1 è richiesto per la fissione perossisomale e suggerendo che il trattamento con SFN provoca fenotipi mitocondriali e perossisomali interrompendo il meccanismo di fissione.
La figura 2 SFN induce l'allungamento perossisomiale. (A) Le cellule RPE-1 sono state trasfettate con 10 nM del siRNA indicato e 3 giorni dopo sono state trattate con DMSO o 50? M SFN per 4 ore. I perossisomi (verdi) sono stati marcati con un anticorpo anti-PMP70, i mitocondri con MitoTracker (rosso) e il DNA controcolorato con DAPI. Gli inserti ingranditi dei perossisomi sono mostrati sulla destra (pannelli d, h e l) per facilitare la visualizzazione dei cambiamenti nella morfologia indotti dall'esaurimento di SFN e Drp1. Le punte delle frecce evidenziano i punti di costrizione. (Per l'interpretazione dei riferimenti al colore nella legenda di questa figura, si rimanda il lettore alla versione web di questo articolo).
Successivamente abbiamo determinato in che modo SFN limita la funzione Drp1. Le possibilità includevano riduzioni dei livelli di espressione, reclutamento / ritenzione ai mitocondri, oligomerizzazione o attività enzimatica della GTPasi. Un deficit in uno qualsiasi di questi comporterebbe una ridotta fissione e iperfusione mitocondriale. Non abbiamo rilevato cambiamenti riproducibili nei livelli di proteina Drp1 dopo il trattamento con SFN (Fig. 1C e 3A), e quindi abbiamo concluso che SFN non altera la stabilità o l'espressione di Drp1, coerente con Drp1 che ha un'emivita> 10 h [50] ei nostri trattamenti SFN sono di durata inferiore. Successivamente, abbiamo studiato se SFN ha influenzato il reclutamento o la ritenzione di Drp1 nei mitocondri. Studi di frazionamento hanno mostrato che SFN ha indotto una perdita di Drp1 dalla frazione mitocondriale (Fig. 3A, corsie 7 e Fig. 8B). Come riportato in precedenza [3], solo una frazione minore di Drp43 (~ 1%) è associata alla rete mitocondriale in un dato momento durante condizioni di stato stazionario con la maggior parte dell'enzima residente nel citoplasma (Fig. 3A, corsie 3 5 ). Questi dati di frazionamento sono stati confermati utilizzando l'analisi di co-localizzazione che ha mostrato una riduzione di ~ 8% nei focolai Drp40 puntati e localizzati nei mitocondri dopo il trattamento con SFN (Fig. 1C e D). Insieme, questi dati indicano che la fusione mitocondriale indotta da SFN è, almeno in parte, dovuta all'associazione attenuata di Drp3 con i mitocondri. I nostri dati non distinguono se SFN interferisce con il reclutamento mitocondriale rispetto alla ritenzione mitocondriale di Drp1, o entrambi, poiché l'analisi del Drp1 endogeno non era suscettibile di visualizzare la GTPasi mediante microscopia a cellule vive.
La figura 3 SFN causa una perdita di Drp1 dai mitocondri. (A) Frazionamento subcellulare di cellule RPE-1 dopo 4 h di DMSO o SFN. I lisati di cellule intere (WCL), nucleari (Nuc), citosolici (Cyto) e frazioni mitocondriali (Mito) sono stati risolti mediante SDS-PAGE e trattati per western blotting con gli anticorpi indicati. La migrazione dei marcatori di peso molecolare è indicata a sinistra. (B) Grafici che mostrano la quantificazione densitometrica di Drp1 nelle frazioni indicate da (A). (C) Le cellule RPE-1 sono state transfettate con 10 nM siCON o siDrp1 e 3 giorni dopo trattati con DMSO o SFN per 4 h. Drp1 (verde) è stato visualizzato con un anticorpo anti-Drp1, mitocondri con MitoTracker (rosso) e nuclei con DAPI (blu). (D) Analisi di co-localizzazione automatica di Drp1 e segnale MitoTracker da (C). I dati in (B) e (D) sono stati compilati dagli esperimenti indipendenti 3 e 5, rispettivamente, e la significatività statistica è stata determinata dal test t di Student a due code. Le barre di errore si riflettono +/- SD e gli asterischi indicano la significatività statistica. (Per l'interpretazione dei riferimenti al colore in questa legenda figura, il lettore è riferito alla versione web di questo articolo).
Sulforaphane conferisce protezione contro l'apoptosi indotta da Staurosportina Indipendente da Nrf2
Lavori precedenti hanno dimostrato che la fissione mitocondriale è permissiva nella formazione dei pori nella membrana mitocondriale esterna generata da Bax / Bak durante l'apoptosi [11]. Drp1 ha dimostrato di essere reclutato selettivamente nei mitocondri durante l'apoptosi [11] e, coerentemente con questo, mitocondri frammentati sono stati osservati all'inizio del processo [27]. Al contrario, si ritiene che l'inibizione della fissione mitocondriale inibisca l'apoptosi bloccando la formazione dei pori della membrana esterna che consentono il rilascio del citocromo c [53]. Di conseguenza, la stimolazione della fusione mitocondriale ritarda la progressione dell'apoptosi indotta da composti tra cui la staurosporina (STS) [14]. Per determinare se SFN protegge le cellule RPE-1 dall'apoptosi mediata da STS e, in tal caso, se ciò richiede Nrf2, abbiamo stabilito un saggio per indurre prontamente la scissione della poli ADP ribosio polimerasi (PARP), un substrato di caspasi-3 attivata e marcatore definitivo di apoptosi. Il trattamento delle cellule RPE-1 con STS 1 M per 6 h ha causato solo una scissione molto modesta di PARP, ma ciò è stato prevenuto dal co-trattamento SFN (ad esempio, Fig. 4A, corsia 3 contro 4). Per aumentare la robustezza di questo test, abbiamo ulteriormente sensibilizzato le cellule all'apoptosi indotta da STS pretrattandole con siRNA mirato al fattore anti-apoptotico, Bcl-XL. Questo pretrattamento ha ridotto l'espressione di Bcl-XL e ha promosso notevolmente la scissione PARP in funzione del tempo esposto a STS (Fig. 4B, confrontare la corsia 2 con le corsie 4-10). È importante sottolineare che 2 ore di pretrattamento con SFN hanno mitigato la scissione di PARP nelle cellule esposte a STS (Fig. 4C, corsia 3 contro 4 e corsia 5 contro 6). Allo stesso modo, le cellule stabilmente impoverite di Nrf2 da CRISPR / Cas9 sono state protette in modo comparabile dalla tossicità STS dal pretrattamento SFN (Fig. 4C, corsia 11 contro 12 e corsia 13 contro 14 e Fig. 4D). Questa protezione è stata osservata utilizzando sia la scissione PARP (Fig. 4C e D) che la morfologia cellulare (Fig. 4E) come letture. L'efficacia dell'esaurimento di Nrf2 da parte di CRISPR / Cas9 è stata confermata mediante western blotting (Fig. 4C, Nrf2 blot). Come previsto, le cellule di deplezione di Drp1, che produce anche un fenotipo di iperfusione (Fig. 1A), hanno anche bloccato la scissione di PARP in risposta a STS rispetto alle cellule di controllo incubate con SFN (Fig. 4F e G). Insieme, questi risultati sono coerenti con SFN che conferisce protezione contro l'apoptosi attraverso la sua capacità di limitare la funzione di Drp1, indipendentemente dalla stabilizzazione e attivazione di Nrf2.
Figura 4 Gli effetti citoprotettivi di SFN sono indipendenti dall'espressione di Nrf2 (A) Le cellule RPE-1 sono state pretrattate con DMSO o 50? M SFN per 2 ore prima del trattamento con DMSO, 1? M staurosporina (STS) o 50? M etoposide per 6 ore e sono stati processati per western blotting anti-PARP. (B) Le cellule RPE-1 sono state trasfettate con 2.5 nM siCON, 1 nM siBcl-XL o 2.5 nM siBcl-XL e 3 giorni dopo sono state trattate con DMSO o 1? M STS per 2, 4 o 6 ore. Vengono mostrati i western blot rappresentativi e la migrazione dei marcatori di peso molecolare è indicata a sinistra. (C) Le cellule RPE-9 generate da CRISPR / Cas2 (Nrf2WT) e knockout Nrf2 (Nrf1KO) sono state trasfettate con 1 nM siBcl-XL e 3 giorni dopo sono state pretrattate con DMSO o 50? M SFN per 2 ore . Successivamente, le cellule sono state trattate con 1? M STS per 2, 4 o 6 ore. Vengono mostrati i western blot rappresentativi con gli anticorpi indicati. (D) Quantificazione del PARP scisso come percentuale del PARP totale (scisso + non tagliato) da 3 esperimenti indipendenti. È importante sottolineare che i livelli di PARP scissa erano comparabili indipendentemente dal fatto che le cellule esprimessero Nrf2 o meno, indicando che la protezione SFN da STS è indipendente dal fattore di trascrizione. (E) Immagini a contrasto di fase 20X scattate immediatamente prima della raccolta dei lisati da (C). Barra della scala = 65 m. (F) Western blot rappresentativi che dimostrano che l'esaurimento di Drp1 conferisce una protezione quasi comparabile da STS come trattamento SFN. Le cellule RPE-1 sono state trasfettate con siBcl-XL da 1 nM e inoltre trasfettate con siCON da 10 nM o siDrp10 da 1 nM. 3 giorni dopo, le cellule siCON sono state pretrattate con SFN come in (A) e (C) e quindi esposte a STS per 4 ore prima di essere raccolte e processate per il western blot con gli anticorpi indicati. (G) Uguale a (D) per i dati presentati in (F) compilati da 3 esperimenti indipendenti. Le barre di errore riflettono +/- SEM
Discussione
Abbiamo scoperto che SFN modula la dinamica di fissione / fusione mitocondriale indipendentemente dai suoi effetti sulla via KEAP1-Nrf2-ARE. Questo è intrigante a causa di un presunto legame tra disfunzione mitocondriale e produzione di ROS e la necessità di squelchare i radicali liberi derivati dai mitocondri attraverso l'attivazione di Nrf2. Questo ulteriore impatto funzionale di SFN è di potenziale importanza dato il numero di studi clinici 30 attualmente in corso per testare SFN per il trattamento di una varietà di malattie tra cui il cancro alla prostata, la broncopneumopatia ostruttiva e l'anemia falciforme [7], [10], [ 47].
Poiché SFN è un isotiocianato [56] e attiva la segnalazione Nrf2 mediante acilazione diretta delle cisteine KEAP1 critiche per sopprimere la degradazione Nrf2 [21], ne consegue che SFN esercita i suoi effetti pro-fusione modulando l'attività di un fattore di fissione o fusione tramite la modificazione della cisteina . I nostri dati sostengono fortemente che Drp1 è regolato negativamente da SFN, anche se resta da chiarire se GTPase sia un obiettivo diretto di acilazione. Nonostante questo divario di conoscenze, la funzione di Drp1 è chiaramente compromessa dall'SFN in quanto sia i mitocondri che i perossisomi diventano iperfusi in risposta al trattamento con SFN e questi organelli condividono Drp1 per i rispettivi eventi di scissione [38]. Inoltre, l'SFN riduce la quantità di Drp1 che si localizza e si accumula nei mitocondri (Fig. 3). Poiché i nostri esperimenti sono stati condotti con tutte le proteine endogene, la nostra scoperta di Drp1 nei siti di fissione mitocondriale è in condizioni di stato stazionario e, di conseguenza, non possiamo distinguere tra un reclutamento e un difetto di ritenzione dell'enzima causato da SFN. Inoltre, non possiamo eliminare la possibilità che gli SFN acilino un recettore ai mitocondri (Fis1 o Mff) per bloccare il reclutamento di Drp1, sospettiamo che Drp1 sia stato modificato direttamente. Drp1 ha nove cisteine, otto delle quali risiedono nel dominio centrale richiesto per l'oligomerizzazione [3], e una delle quali risiede nel dominio GTPase Effector (GED) al C-terminale di Drp1. L'acilazione diretta di una qualsiasi di queste cisteine potrebbe causare un difetto di attività in Drp1 e quindi essere alla base dell'effetto di SFN sulla dinamica mitocondriale. In particolare, il lavoro precedente suggerisce che i difetti nell'oligomerizzazione e l'attività catalitica possono abrogare la ritenzione di Drp1 nei mitocondri [52]. Cys644 nel dominio GED è un obiettivo particolarmente attraente basato su un lavoro precedente che mostra la mutazione di queste mutazioni di cisteina che alterano l'attività di Drp1 GTPase [4] e che questa particolare cisteina è modificata da elettrofili reattivi al tiolo [9]. La risoluzione di questa domanda in sospeso richiederà la convalida spettrometrica di massa. In sintesi, abbiamo identificato una nuova funzione citoprotettiva per il composto SFN clinicamente rilevante. Oltre ad attivare il principale fattore di trascrizione antiossidante Nrf2, SFN promuove la fusione mitocondriale e perossisomale e questo effetto è indipendente da Nrf2. Il meccanismo alla base di questo fenomeno comporta una riduzione della funzione della GTPasi Drp1, il mediatore primario della fissione mitocondriale e perossisomale. Una delle principali conseguenze della fusione mitocondriale mediata da SFN è che le cellule diventano resistenti agli effetti tossici dell'induttore di apoptosi staurosporina. Questa ulteriore azione citoprotettiva di SFN potrebbe essere di particolare utilità clinica nelle numerose malattie neurodegenerative per le quali l'età è il principale fattore di rischio (ad esempio, morbo di Parkinson, morbo di Alzheimer, degenerazione maculare legata all'età) poiché tali malattie sono state associate all'apoptosi e ridotto livelli e / o disregolazione di Nrf2 [35], [36], [48].
Materiali e Metodi
Saggi di apoptosi
Le cellule sono state seminate e trasfettate con siRNA come indicato di seguito. Le cellule sono state pretrattate con 50 µM di sulforafano per 2 h per indurre la fusione mitocondriale e poi sono state trattate con 1 µM di staurosporina per indurre l'apoptosi. Al momento della raccolta, i terreni sono stati raccolti in singole provette e sottoposti a centrifugazione ad alta velocità per ottenere cellule apoptotiche a pellet. Questo pellet cellulare è stato combinato con cellule aderenti e solubilizzato in tampone Laemmli concentrato 2 volte. I campioni sono stati sottoposti a western blot anti-PARP.
Generazione di costruzione CRISPR / Cas9
Per creare LentiCRISPR / eCas9 1.1, LentiCRISPR v2 (aggiunta #52961) è stato dapprima tagliato con Age1 e BamH1. Successivamente, SpCas9 da eSpCas9 1.1 (addgene #71814) è stato amplificato con PCR amplificato con Age1 e BamH1 utilizzando i seguenti primer (avanti AGCGCACCGGTTCTAGAGCGCTGCCACCATGGACTATAAGGACCACGAC, Reverse AAGCGCGGATCCCTTTTTCTTTTTTGCCTGGCCGG) e ligato nel vettore di taglio sopra. sequenze di sgRNA sono state determinate utilizzando Benchling.com. I parametri sono stati impostati per indirizzare la sequenza di codifica con il punteggio più alto sul bersaglio e il più basso fuori bersaglio. Le seguenti sequenze (sequenza target sottolineata, hs sgNFE2L2 # 1 senso CACCGCGACGGAAAGAGTATGAGC, antisenso AAACGCTCATACTCTTTCCGTCGC; hs sgNFE2L2 # 2 senso CACCGGTTTCTGACTGGATGTGCT, antisenso AAACAGCACATCCAGTCAGAAACC; hs sgNFE2L2 # 3 senso CACCGGAGTAGTTGGCAGATCCAC, antisenso AAACGTGGATCTGCCAACTACTCC) sono stati ricotto e ligati in BsmB1 tagliato LentiCRISPR / eCas9 1.1. Le cellule RPE-1 infettate con lentivirus sono state selezionate con puromicina e mantenute come una popolazione aggregata. Knockout è stato confermato da immunofluorescenza e western blotting.
Coltura cellulare e trasfezione
Cellule epiteliali pigmentate retiniche umane trasformate con telomerasi (RPE-1) (ATCC) sono state coltivate in Dulbecco's Modified Eagle Medium (DMEM) contenente 1 g / L di glucosio integrato con penicillina, streptomicina, cocktail di amminoacidi non essenziali 1X (Life Technologies), e 10% di siero bovino fetale (Life Technologies). Per le trasfezioni di siRNA, 30,000 35,000 cellule / mL sono state seminate durante la notte. Le cellule hanno ricevuto 10 nM di siRNA diluito in DMEM privo di siero e combinato con il reagente di trasfezione dell'interferina allo 0.3% (PolyPlus). Per la sensibilizzazione all'apoptosi, le cellule hanno ricevuto siRNA Bcl-XL da 1 nM. Le cellule sono state raccolte 2-3 giorni dopo la trasfezione.
Prodotti chimici, anticorpi e siRNA Oligos
Anticorpi contro? -Tubulina (Segnalazione cellulare),? -Tubulina (Sigma), Drp1 (BD Biosciences), KEAP1 (Proteintech), Lamin B1 (Abcam), PARP (Segnalazione cellulare), PMP70 (Abcam) e Tom20 (BD Biosciences ) sono stati usati a diluizioni 1: 1000 per il western blotting e per l'immunofluorescenza. In-house, l'anticorpo di coniglio anti-Nrf2 è stato utilizzato a 1: 2000 per il western blotting [34], [59]. Sulforafano (Sigma) e staurosporina (Tocris) sono stati utilizzati rispettivamente a 50 µM e 1 µM. siRNA contro Drp1 (Dharmacon), Nrf2 (Dharmacon), KEAP1 (Segnalazione cellulare) e Bcl-XL (Segnalazione cellulare) sono stati utilizzati a 10 nM se non diversamente specificato.
Immunofluorescenza e in Vivo Labeling
Le cellule seminate su coprioggetti di vetro da 18 mm sono state trattate con veicolo o farmaco, fissate in formaldeide al 3.7% e quindi permeabilizzate in Triton X-0.2 / PBS allo 100% su ghiaccio per 10 min. Gli anticorpi primari sono stati incubati in albumina sierica bovina al 3% (BSA) in PBS per una notte a 4 ° C. Dopo i lavaggi PBS, le cellule sono state incubate per 1 ora in anticorpi secondari coniugati Alexa488- o Alexa546- adatti alla specie (diluiti 1: 1000) e 0.1 μg / mL DAPI (Sigma) in 3% BSA / PBS. I mitocondri sono stati visualizzati mediante immunofluorescenza anti-Tom20 o incubando cellule in 200 nM MitoTracker Red CMXRos (Molecular Probes, Inc.) in DMEM senza siero per 30 min a 37 ° C prima della fissazione.
Microscopia e analisi delle immagini
I campioni di immunofluorescenza sono stati visualizzati su un microscopio confocale LSM710 (Carl Zeiss). Le micrografie sono state acquisite utilizzando obiettivi a immersione in olio 63X o 100X e le immagini sono state regolate e migliorate utilizzando Adobe Photoshop CS6. L'analisi di co-localizzazione è stata eseguita utilizzando la funzione di co-localizzazione Carl Zeiss LSM710 con soglie impostate manualmente mentre non si conosceva l'identità dei campioni. Le barre della scala in tutto, se non diversamente indicato, sono 10 m. La morfologia mitocondriale è stata valutata mediante punteggio in cieco. Se i mitocondri di una cellula erano mantenuti come puncta multipli, rotondi, discriminanti, la cellula veniva valutata come "fissazione". Se i singoli mitocondri erano indistinguibili e l'intera rete mitocondriale appariva continua, la cellula veniva classificata come "fusione". Tutte le altre cellule, comprese quelle con mitocondri in cluster, sono state classificate come "intermedie".
Frazionamenti subcellulari
Le cellule RPE-1 sono state coltivate fino alla confluenza. Dopo un lavaggio con PBS, le cellule sono state sottoposte a centrifugazione a 600 g per 10 min e risospese in tampone di isolamento 600 μl (mannitolo 210 mM, saccarosio 70 mM, MOPS 5 mM, EDTA 1 mM pH 7.4 + PMSF 1 mM). La sospensione è stata lisata 30 volte in un omogeneizzatore Dounce. Una frazione dell'omogenato è stata conservata come "lisato cellulare intero". Il resto è stato sottoposto a centrifugazione a 800 g per 10 min sui nuclei del pellet. I surnatanti sono stati sottoposti a centrifugazione a 1500 g per 10 min per eliminare i nuclei rimanenti e le cellule non lisate. Questo supernatante è stato sottoposto a centrifugazione a 15,000 g per 15 min in mitocondri a pellet. Il surnatante è stato conservato come la `` frazione citosolica ''. Il pellet è stato lavato delicatamente con PBS e risospeso in tampone di isolamento. La concentrazione proteica di ciascuna frazione è stata misurata mediante saggio di acido bicinconinico (BCA) e quantità equivalenti di proteine sono state risolte mediante SDS-PAGE.
Macchia occidentale
Le cellule sono state lavate in PBS e solubilizzate in tampone solubilizzante Laemmli concentrato 2 volte (100 mM Tris [pH 6.8], 2% SDS, 0.008% blu di bromofenolo, 2% 2-mercaptoetanolo, 26.3% glicerolo e 0.001% pirinina Y). I lisati sono stati fatti bollire per 5 minuti prima del caricamento su gel di poliacrilammide di sodio dodecil solfato (SDS). Le proteine sono state trasferite alle membrane di nitrocellulosa e le membrane sono state bloccate per 1 ora in 5% di latte / TBST. Gli anticorpi primari sono stati diluiti in 5% di latte / TBST e incubati con il blot per una notte a 4 ° C. Gli anticorpi secondari coniugati con perossidasi di rafano (HRP) sono stati diluiti in 5% di latte / TBST. Le macchie sono state elaborate con chemiluminescenza potenziata e le quantificazioni densitometriche sono state eseguite utilizzando il software ImageJ.
Il sulforafano è una sostanza chimica della raccolta di isotiocianato di sostanze organosolfuro ottenute da verdure crocifere, tra cui broccoli, cavoli, cavolfiori, cavoli e cavoli, tra gli altri. Il sulforafano viene prodotto quando l'enzima mirosinasi trasforma la glucorafanina, un glucosinolato, nel sulforafano, noto anche come sulforaphane-glucosinolato. I germogli di broccoli e il cavolfiore hanno la più alta concentrazione di glucorafanina o il precursore del sulforafano. Studi di ricerca hanno dimostrato che il sulforafano migliora le capacità antiossidanti dell'organismo umano per prevenire vari problemi di salute. Dr. Alex Jimenez DC, CCST Insight
Sulforafano e suoi effetti su cancro, mortalità, invecchiamento, cervello e comportamento, malattie cardiache e altro
Gli isotiocianati sono alcuni dei composti vegetali più importanti che si possono ottenere nella dieta. In questo video faccio il caso più completo per loro che sia mai stato fatto. Soglia di attenzione breve? Passa al tuo argomento preferito facendo clic su uno dei seguenti punti temporali. Timeline completa di seguito.
Sezioni chiave:
00: 01: 14 - Cancro e mortalità
00: 19: 04 - Invecchiamento
00: 26: 30 - Cervello e comportamento
00: 38: 06 - Riassunto finale
00: 40: 27 - Dose
Timeline completa:
00: 00: 34 - Introduzione di sulforaphane, uno degli obiettivi principali del video.
00: 01: 14 - Consumo di verdure crocifere e riduzione della mortalità per tutte le cause.
00: 02: 12 - Rischio di cancro alla prostata.
00: 02: 23 - Rischio di cancro alla vescica.
00: 02: 34 - Carcinoma polmonare nei fumatori.
00: 02: 48 - Rischio di cancro al seno.
00: 03: 13 - Ipotetico: cosa succede se hai già un cancro? (Interventistica)
00: 03: 35 - Meccanismo plausibile che guida i dati associativi sul cancro e sulla mortalità.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Prova animale che mostra un forte effetto dell'estratto di germogli di broccolo sullo sviluppo del tumore della vescica nei ratti.
00: 06: 06 - Effetto dell'integrazione diretta di sulforafano nei pazienti affetti da cancro alla prostata.
00: 07: 09 - Bioaccumulo di metaboliti di isotiocianato nel tessuto mammario effettivo.
00: 08: 32 - Inibizione delle cellule staminali del carcinoma mammario.
00: 08: 53 - Lezione di storia: le brassiche sono state istituite come aventi proprietà sanitarie anche nell'antica Roma.
00: 09: 16 - La capacità del Sulforaphane di potenziare l'escrezione di cancerogeno (benzene, acroleina).
00: 09: 51 - NRF2 come interruttore genetico tramite elementi di risposta antiossidante.
00: 10: 10 - Come l'attivazione di NRF2 migliora l'escrezione di cancerogeno tramite glutatione-S-coniugati.
00: 10: 34 - I cavoletti di Bruxelles aumentano la glutatione-S-transferasi e riducono il danno al DNA.
00: 11: 20 - La bevanda di germogli di broccoli aumenta l'escrezione di benzene di 61%.
00: 13: 31 - L'omogenato di germogli di broccoli aumenta gli enzimi antiossidanti nelle vie aeree superiori.
00: 15: 45 - Consumo di verdure crocifere e mortalità per malattie cardiache.
00: 16: 55 - La polvere di germogli di broccoli migliora i lipidi nel sangue e il rischio complessivo di malattie cardiache nei diabetici di tipo 2.
00: 19: 04 - Inizio della sezione di invecchiamento.
00: 19: 21 - La dieta arricchita con sulforafano migliora la durata della vita dei coleotteri da 15 a 30% (in determinate condizioni).
00: 20: 34 - L'importanza della bassa infiammazione per la longevità.
00: 22: 05 - Le verdure crocifere e la polvere di germogli di broccoli sembrano ridurre un'ampia varietà di marcatori infiammatori negli esseri umani.
00: 23: 40 - Ricapitolazione di metà video: cancro, sezioni di invecchiamento
00: 24: 14 - Gli studi sui topi suggeriscono che il sulforafano potrebbe migliorare la funzione immunitaria adattativa in età avanzata.
00: 25: 18 - Sulforaphane ha migliorato la crescita dei peli in un modello murino di calvizie. Immagine su 00: 26: 10.
00: 26: 30 - Inizio della sezione cervello e comportamento.
00: 27: 18 - Effetto dell'estratto di germogli di broccoli sull'autismo.
00: 27: 48 - Effetto del glucorapanin sulla schizofrenia.
00: 28: 17 - Inizio della discussione sulla depressione (meccanismo e studi plausibili).
00: 31: 21 - Lo studio del mouse utilizzando 10 diversi modelli di depressione indotta da stress mostra sulforapano in modo simile efficace come fluoxetina (prozac).
00: 32: 00 - Lo studio mostra che l'ingestione diretta di glucorafanina nei topi è altrettanto efficace nel prevenire la depressione dal modello di stress sociale di sconfitta.
00: 33: 01 - Inizio della sezione di neurodegenerazione.
00: 33: 30 - Sulforaphane e malattia di Alzheimer.
00: 33: 44 - Sulforaphane e morbo di Parkinson.
00: 33: 51 - Sulforaphane e la malattia di Hungtington.
00: 34: 43 - Inizio della sezione traumatica di lesioni cerebrali.
00: 35: 01 - Sulforaphane iniettato immediatamente dopo TBI migliora la memoria (studio del mouse).
00: 35: 55 - Sulforaphane e plasticità neuronale.
00: 36: 32 - Sulforaphane migliora l'apprendimento nel modello di diabete di tipo II nei topi.
00: 37: 19 - Distrofia muscolare sulforapano e duchenne.
00: 37: 44 - Inibizione della miostatina nelle cellule muscolari satelliti (in vitro).
00: 38: 06 - Ricapitolazione tardiva: mortalità e cancro, danno al DNA, stress ossidativo e infiammazione, escrezione di benzene, malattie cardiovascolari, diabete di tipo II, effetti sul cervello (depressione, autismo, schizofrenia, neurodegenerazione), via NRF2.
00: 40: 27 - Pensieri sulla determinazione di una dose di germogli di broccoli o sulforafano.
00: 41: 01 - Aneddoti su germinazione a casa.
00: 43: 14 - Sulle temperature di cottura e sull'attività del sulforafano.
00: 43: 45 - Conversione batterica intestinale del sulforafano da glucorafanina.
00: 44: 24 - Gli integratori funzionano meglio se combinati con la mirosinasi attiva delle verdure.
00: 44: 56 - Tecniche di cottura e verdure crucifere.
Il riscaldamento diminuisce l'attività delle proteine epitelio-magiche e aumenta la formazione di sulforafano nei broccoli
Astratto
Il sulforafano, un isotiocianato dei broccoli, è uno dei più potenti anticancerogeni di derivazione alimentare. Questo composto non è presente nel vegetale intatto, piuttosto è formato dal suo precursore glucosinolato, la glucorafanina, per azione della mirosinasi, un enzima tioglucosidasi, quando il tessuto dei broccoli viene frantumato o masticato. Tuttavia, numerosi studi hanno dimostrato che la resa di sulforafano dalla glucorafanina è bassa e che un analogo del nitrile non bioattivo, il sulforafano nitrile, è il principale prodotto di idrolisi quando il tessuto vegetale viene frantumato a temperatura ambiente. Prove recenti suggeriscono che in Arabidopsis, la formazione di nitrile dai glucosinolati è controllata da una proteina sensibile al calore, la proteina epithiospecifier (ESP), un cofattore non catalitico della mirosinasi. I nostri obiettivi erano esaminare gli effetti del riscaldamento di cimette e germogli di broccoli sulla formazione di sulforafano e nitrile di sulforafano, per determinare se i broccoli contengono attività ESP, quindi correlare i cambiamenti dipendenti dal calore nell'attività ESP, contenuto di sulforafano e bioattività, misurata mediante induzione del enzima di disintossicazione di fase II chinone reduttasi (QR) in coltura cellulare. Il riscaldamento di cimette di broccoli freschi o germogli di broccoli a 60 ° C prima dell'omogeneizzazione aumenta contemporaneamente la formazione di sulforafano e diminuisce la formazione di nitrile di sulforafano. Una significativa perdita di attività ESP è stata parallela alla diminuzione della formazione di nitrile di sulforafano. Il riscaldamento a 70 ° C e oltre ha ridotto la formazione di entrambi i prodotti nei fiori di broccoli, ma non nei germogli di broccoli. L'induzione del QR nelle cellule di epatoma Hepa lclc7 di topo in coltura ha parallelamente l'aumento della formazione di sulforafano.
Il preriscaldamento di cimette e germogli di broccoli a 60 ° C ha aumentato significativamente la formazione catalizzata dalla mirosinasi di sulforafano (SF) negli estratti di tessuto vegetale dopo la frantumazione. Ciò è stato associato a una diminuzione della formazione di nitrile di sulforafano (nitrile SF) e dell'attività della proteina epitospecificante (ESP).
In conclusione, il sulforafano è un fitochimico presente nei broccoli e in altre verdure crocifere. Una quantità incontrollata di ossidanti causata da fattori sia interni che esterni può causare stress ossidativo nel corpo umano che alla fine può portare a una varietà di problemi di salute. Il sulforafano può attivare la produzione di Nrf2, un fattore di trascrizione che aiuta a regolare i meccanismi antiossidanti protettivi che controllano la risposta della cellula agli ossidanti. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
Discussione aggiuntiva sull'argomento: Dolore alla schiena acuto
Mal di schienaè una delle cause più diffuse di disabilità e di giornate di lavoro perse in tutto il mondo. Il dolore alla schiena si attribuisce alla seconda ragione più comune per le visite mediche, superata solo dalle infezioni delle alte vie respiratorie. Circa l'80% della popolazione sperimenterà dolore alla schiena almeno una volta nella vita. La colonna vertebrale è una struttura complessa composta da ossa, articolazioni, legamenti e muscoli, tra gli altri tessuti molli. Per questo motivo, lesioni e / o condizioni aggravate, come dischi erniciati, può eventualmente portare a sintomi di mal di schiena. Le lesioni sportive o gli incidenti automobilistici sono spesso la causa più frequente di mal di schiena, tuttavia a volte il più semplice dei movimenti può avere risultati dolorosi. Fortunatamente, le opzioni di trattamento alternative, come la cura chiropratica, possono aiutare ad alleviare il mal di schiena attraverso l'uso di aggiustamenti spinali e manipolazioni manuali, in definitiva migliorando il sollievo dal dolore.
Lo strumento Find A Practitioner di IFM è la più grande rete di riferimento in Medicina Funzionale, creata per aiutare i pazienti a localizzare professionisti di Medicina Funzionale in qualsiasi parte del mondo. I professionisti certificati IFM sono elencati per primi nei risultati di ricerca, data la loro vasta formazione in Medicina Funzionale
Prenotazione online e appuntamenti 24 ore su 7, XNUMX giorni su XNUMX *