Le malattie neurodegenerative, come il morbo di Alzheimer e il morbo di Parkinson, colpiscono milioni di persone in tutto il mondo. Sono disponibili varie opzioni di trattamento per trattare i sintomi di diverse malattie neurodegenerative, sebbene i risultati siano spesso limitati. Studi di ricerca hanno trovato che lo stress ossidativo causato da fattori sia interni che esterni può essere una causa per lo sviluppo di malattie neurodegenerative. Il fattore di trascrizione, Nrf2, è stato determinato a funzionare come un importante meccanismo di difesa contro lo stress ossidativo. Lo scopo dell'articolo qui sotto è quello di mostrare gli effetti di Nrf2 su malattie neurodegenerative.
Contenuti
Modulazione di Proteostasi mediante Fattore di trascrizione NRF2
Le malattie neurodegenerative sono legate all'accumulo di specifici aggregati proteici, suggerendo una connessione intima tra il cervello ferito e la perdita di proteostasi. La proteostasi si riferisce a tutti i processi attraverso i quali le cellule controllano l'abbondanza e il ripiegamento del proteoma grazie ad un'ampia rete che integra la regolazione delle vie di segnalazione, dell'espressione genica e dei sistemi di degradazione delle proteine. Questa recensione tenta di riassumere i risultati più rilevanti sulla modulazione trascrizionale della proteostasi esercitata dal fattore di trascrizione NRF2 (fattore 2 simile a un fattore nucleare (2 derivato da eritroidi). NRF2 è stato classicamente considerato il principale regolatore della risposta delle cellule antiossidanti, sebbene attualmente stia emergendo come componente chiave dei macchinari di trasduzione per mantenere la proteostasi. Come discuterà, NRF2 potrebbe essere immaginato come un hub che compila segnali di emergenza derivati dall'accumulazione di proteine misfolded al fine di costruire una risposta trascrizionale coordinata e perdurabile. Ciò è ottenuto dalle funzioni di NRF2 relative al controllo dei geni coinvolti nel mantenimento della fisiologia del reticolo endoplasmatico, del proteasoma e dell'autofagia.
parole chiave: Malattie neurodegenerative, risposta proteica non spiegata, proteasoma, ubiquitina, autofagia, stress ossidativo
Abbreviazioni
Sciencedirect.com/science/article/pii/S2213231716304050
Introduzione
Fattore Nucleare (2 derivato da eritroide) 2 (NRF2) è una proteina base-leucina-cerniera considerata al giorno d'oggi come un maestro regolatore dell'omeostasi cellulare. Controlla l'espressione basale e stress-inducibile di oltre i geni 250 che condividono in comune un potenziatore cis-acting denominato elemento di risposta antiossidante (ARE) [1], [2], [3], [4], [5]. Questi geni partecipano alle reazioni di disintossicazione di fase I, II e III, al glutatione e al metabolismo di peroxiredoxin / thioredoxin, alla produzione di NADPH attraverso la via del pentoso fosfato e l'enzima malico, l'ossidazione degli acidi grassi, il metabolismo del ferro e la proteostasi [6]. Date queste ampie funzioni citoprotettive, è possibile che un singolo colpo farmacologico in NRF2 possa mitigare l'effetto dei principali colpevoli di malattie croniche, tra cui lo stress ossidativo, infiammatorio e proteotossico. Il ruolo di NRF2 nella modulazione della difesa antiossidante e la risoluzione dell'infiammazione sono stati affrontati in numerosi studi (recensione in [7]). Qui, ci concentreremo sul suo ruolo nella proteostasi, cioè il controllo omeostatico della sintesi proteica, del ripiegamento, del traffico e della degradazione. Verranno forniti esempi nel contesto delle malattie neurodegenerative.
La perdita di proteostasi influenza l'attività di NRF2 nelle malattie neurodegenerative
Un segno distintivo generale delle malattie neurodegenerative è il verificarsi di aggregazioni aberranti di alcune proteine. Pertanto, aggregati proteici mal ripiegati di? -Sinucleina (? -SYN) si trovano nelle placche del morbo di Parkinson (PD),? -Amiloide (A?) E grovigli neurofibrillari TAU iperfosforilati nella malattia di Alzheimer (AD), huntingtina (Htt) in Malattia di Huntington (HD), superossido dismutasi 1 (SOD1) e proteina legante il DNA di TAR 43 (TDP-43) nella sclerosi laterale amiotrofica (SLA), proteina prionica (PrP) nelle encefalopatie spongiformi, ecc. Gli aggregati proteici possono avere un impatto su diversi percorsi cellulari, che a loro volta possono influenzare i livelli e l'attività di NRF2.
Diversi livelli di regolazione controllano strettamente l'attività di NRF2
In condizioni fisiologiche, le cellule mostrano bassi livelli di proteina NRF2 a causa del suo rapido turnover. In risposta a diversi stimoli, la proteina NRF2 viene accumulata, entra nel nucleo e aumenta la trascrizione dei geni contenenti ARE. Pertanto, la gestione dei livelli di proteina NRF2 è un punto chiave che dovrebbe integrare segnali di input positivi e negativi. Come discuterà ulteriormente, NRF2 è attivato da diversi meccanismi sovrapposti per orchestrare una risposta rapida ed efficiente ma d'altra parte NRF2 potrebbe essere inibito, probabilmente in una seconda fase, al fine di disattivare la sua risposta.
Dal punto di vista classico, l'attivazione di NRF2 è stata considerata come una conseguenza della risposta cellulare a composti ossidanti o elettrofili. A questo proposito, l'adattatore della ligasi E3 dell'ubiquitina, la proteina 1 associata a Kelch-like ECH (KEAP1) gioca un ruolo cruciale. I dettagli molecolari saranno ulteriormente trattati nella Sezione 4.1. In breve, KEAP1 agisce come un sensore redox a causa dei residui critici di cisteina che portano all'ubiquitinazione di NRF2 e alla degradazione proteasomica. Oltre a questa classica modulazione, NRF2 è profondamente regolato dalla segnalazione di eventi. In effetti, è stato dimostrato che diverse chinasi fosforilano e regolano NRF2. Ad esempio, NRF2 può essere fosforilato dalle protein chinasi attivate dal mitogeno (MAPK), sebbene il suo contributo all'attività di NRF2 rimanga poco chiaro [8], [9], [10], [11]. Chinasi PKA così come alcuni isoenzimi PKC [12], CK2 [13] o Fyn [14] fosforilano NRF2 modificandone la stabilità. Un precedente lavoro del nostro gruppo ha riportato che glicogeno sintasi kinse-3? (GSK-3?) Inibisce NRF2 per esclusione nucleare e degradazione proteasomiale [15], [25], [26], [27], [28], [29], [30]. I dettagli molecolari saranno discussi nella Sezione 4.1. Inoltre, NRF2 è sottoposto ad altri tipi di regolamentazione. Ad esempio, l'acetilazione di NRF2 da parte di CBP / p300 aumenta la sua attività [17], mentre è inibita da miR153, miR27a, miR142-5p e miR144 [16] o dalla metilazione delle isole citosina-guanina (CG) all'interno del promotore NRF2 [18].
Impatto degli aggregati proteici sui meccanismi regolatori di NRF2
In questa sezione ci concentreremo su come l'accumulo di proteine misfolded potrebbe avere un impatto sull'attività di NRF2 fornendo alcuni dei percorsi sopra menzionati come esempi illustrativi. In primo luogo, dobbiamo considerare che l'accumulo di proteine è strettamente legato al danno ossidativo. Infatti, l'accumulo e l'aggregazione di proteine mal ripiegate inducono una produzione anormale di specie reattive dell'ossigeno (ROS) dai mitocondri e da altre fonti [19]. Come accennato in precedenza, ROS modificherà le cisteine sensibili al redox di KEAP1 portando al rilascio, stabilizzazione e localizzazione nucleare di NRF2.
Per quanto riguarda le proteinopatie, un esempio di eventi di segnalazione disregolati che possono influenzare NRF2 è fornito dall'iperattivazione di GSK-3? in AD. GSK-3?, Noto anche come chinasi TAU, partecipa alla fosforilazione di questa proteina associata ai microtubuli, con conseguente aggregazione, formazione di grovigli neurofibrillari e interruzione del trasporto assonale (rivisto in [20]). D'altra parte, GSK-3? riduce drasticamente i livelli e l'attività di NRF2 come menzionato sopra. Sebbene non ampiamente accettata, la cascata dell'amiloide propone che tossico A? gli oligomeri aumentano GSK-3? attività insieme all'iperfosforilazione della TAU e alla morte dei neuroni [21], [22]. Esistono diversi modelli per spiegare come A? favorisce GSK3-? attività. Ad esempio, A? si lega al recettore dell'insulina e inibisce le vie di segnalazione PI3K e AKT, che sono cruciali per mantenere GSK-3? inattivato dalla fosforilazione nel suo residuo Ser9 N-terminale [23]. D'altra parte, extracellulare A? interagisce con i recettori Frizzled, bloccando la segnalazione WNT [24] e di nuovo determinando il rilascio di GSK-3 attivo ?. In sintesi, A? l'accumulo porta ad un'iperattivazione anormale di GSK-3?, compromettendo così un'appropriata risposta NRF2.
Come discusso nella sezione seguente, le proteine mal ripiegate portano all'attivazione di PERK e MAPK, che a loro volta regolano su NRF2 [31], [8], [9], [10], [11]. Inoltre, l'attività CBP / p300 disregolata è stata riportata in diverse proteinopatie [32] e una diminuzione globale della metilazione del DNA nei cervelli AD è stata anche dimostrata [33], fornendo quindi motivi per esplorare la rilevanza di questi risultati nel regolamento NRF2.
Noi e altri abbiamo osservato in necroscopie di pazienti con PD e AD un aumento dei livelli di proteina NRF2 e alcuni dei suoi bersagli, come l'ossigenoterasi 1 (HMOX1), il NADPH chinone ossidasi 1 (NQO1), p62, ecc., Sia da immunoblot che per immunoistochimica [34], [35], [36], [37], [38], [39]. L'up-regulation di NRF2 in queste malattie è interpretato come un tentativo infruttuoso del cervello malato di recuperare i valori omeostatici. Tuttavia, un altro studio ha indicato che NRF2 è prevalentemente localizzato nel citoplasma dei neuroni dell'ippocampo AD, suggerendo una ridotta attività trascrizionale di NRF2 nel cervello [40]. È concepibile che la disparità di queste osservazioni sia correlata ai cambiamenti nei fattori che controllano NRF2 lungo le fasi progressive della neurodegenerazione.
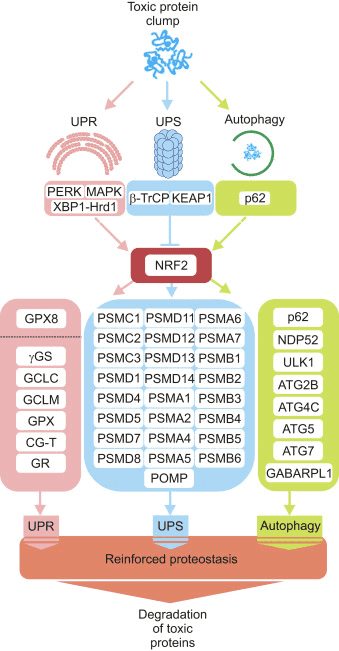
Tre sistemi principali contribuiscono alla proteostasi, vale a dire la risposta proteica dispiegata (UPR), il sistema di proteasoma ubiquitina (UPS) e l'autofagia. Successivamente, presentiamo prove per immaginare NRF2 come un hub che collega segnali di emergenza attivati da aggregati proteici con il meccanismo derivato dalle proteine.
NRF2 partecipa alla risposta proteica non spiegata (UPR)
Attivazione NRF2 in risposta a UPR
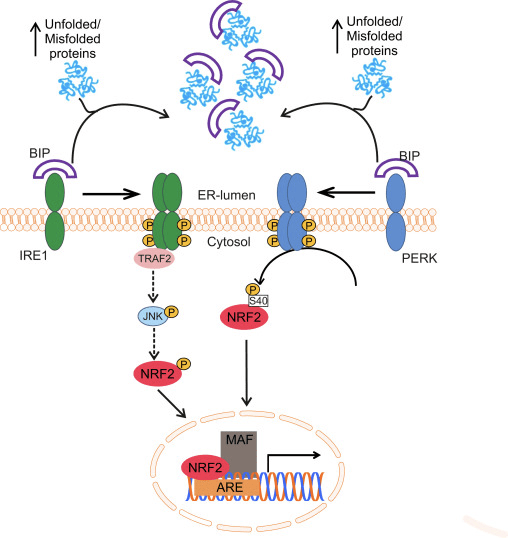
Il ripiegamento delle proteine ossidative nel pronto soccorso è guidato da una serie di percorsi distinti, il più conservato dei quali coinvolge la proteina disolfuro-isomerasi (PDI) e la sulfidril ossidasi ossidoreduttina endoplasmatica 1 (ERO1? E ERO1? Nei mammiferi) come donatore di disolfuro. In breve, la PDI catalizza la formazione e la rottura dei legami disolfuro tra i residui di cisteina all'interno delle proteine, man mano che si ripiegano, a causa della riduzione e dell'ossidazione dei propri amminoacidi cisteina. La PDI viene riciclata dall'azione dell'enzima di mantenimento della casa ERO1, che reintroduce i legami disolfuro nella PDI [41]. L'ossigeno molecolare è l'accettore di elettroni terminale di ERO1, che genera quantità stechiometriche di perossido di idrogeno per ogni legame disolfuro prodotto [42]. Le perossidasi (PRX4) e le perossidasi di glutatione (GPX7 e GPX8) sono enzimi chiave per ridurre il perossido di idrogeno nel pronto soccorso. Quando questo sistema ossidoriduttivo non funziona correttamente, si verifica un accumulo anormale di proteine mal ripiegate nel pronto soccorso e una serie di segnali denominati risposta proteica spiegata (UPR) viene trasmessa al citoplasma e al nucleo per ristabilire l'omeostasi del pronto soccorso [43]. Sono state identificate tre proteine associate alla membrana per rilevare lo stress ER negli eucarioti: fattore di trascrizione attivante 6 (ATF6), ER pancreatico eIF2? chinasi (PERK, anche chinasi ER simile alla proteina chinasi attivata da RNA a doppio filamento) e chinasi1 che richiede inositolo (IRE1). Il dominio luminale di ciascun sensore è legato a una chaperone da 78 kDa chiamata proteina regolata dal glucosio (GRP78 / BIP). Il BIP si dissocia in seguito allo stress ER per legare le proteine dispiegate, portando all'attivazione dei tre sensori [44].
NRF2 e il suo omologo NRF1, anche in relazione alla risposta antiossidante, partecipano alla trasduzione dell'UPR nel nucleo. Nel caso di NRF1, questa proteina si trova nella membrana ER e subisce traslocazione nucleare dopo deglicosilazione o scissione. Quindi, l'attivazione di UPR porta all'elaborazione di NRF1 e all'accumulo nucleare del frammento risultante nel compartimento nucleare. Tuttavia, la capacità di transattivare i geni contenenti ARE di questo frammento NRF1 è ancora in discussione [45].
Glover-Cutter e collaboratori hanno mostrato l'attivazione dell'ortologo NRF2 di C. elegans, SKN-1, con diversi fattori di stress ER. L'aumento dell'espressione di SKN-1 dipendeva da diversi mediatori UPR, inclusi i ortologhi worm IRE1 o PERK [46]. Nelle cellule con carenza di PERK, la sintesi proteica danneggiata porta all'accumulo di perossidi endogeni e conseguente apoptosi [47]. L'effettore utilizzato da PERK per proteggere l'ER da questi perossidi potrebbe essere NRF2, poiché è stato riportato che PERK fosforila NRF2 a Ser40, impedendone così la degradazione da parte di KEAP1 [31]. L'induzione di ASK1 è anche probabile che giochi un ruolo in questa via attraverso l'azione della chinasi mediata da TRAF2 di IRE1 [48]. Sebbene il ruolo dei MAPK nella regolazione di NRF2 sia ancora controverso, è stato recentemente suggerito che il percorso IRE1-TRAF2-ASK1-JNK possa attivare NRF2 [49] (Fig. 1). È interessante notare che in C. elegans e cellule umane, nuove evidenze suggeriscono che la cisteina di solfenilazione della chinasi di IRE1 nel suo ciclo di attivazione inibisce l'UPR mediato da IRE1 e avvia una risposta antiossidante p38 guidata da NRF2. I dati suggeriscono che IRE1 ha un'antica funzione di sentinella citoplasmatica che attiva p38 e NRF2 [50].
Molti studi sull'induzione dell'UPR sono stati condotti con l'inibitore della tunicamicina della glicosilazione proteica. NRF2 sembra essere essenziale per la prevenzione della morte cellulare per apoptosi indotta dalla tunicamicina [31] e la sua attivazione in queste condizioni è guidata dalla degradazione autofagica di KEAP1 [51]. Di conseguenza, il silenziamento mediato da shRNA dell'espressione di NRF2 nelle cellule? TC-6, una linea cellulare insulinoma? Murina, ha aumentato significativamente la citotossicità indotta dalla tunicamicina e ha portato ad un aumento dell'espressione del marker di stress ER pro-apoptotico CHOP10. D'altra parte, l'attivazione di NRF2 da parte dell'1,2-ditiolo-3-tione (D3T) ha ridotto la citotossicità della tunicamicina e attenuato l'espressione di CHOP10 e PERK [52]. È interessante notare che i neuroni olfattivi sottoposti ad applicazione sistemica di tunicamicina hanno aumentato NRF2 in parallelo con altri membri UPR come CHOP, BIP, XBP1 [53]. Questi risultati sono stati estesi a studi in vivo, poiché l'infusione ventricolare laterale di tunicamicina nei ratti ha indotto l'espressione di PERK e NRF2 nell'ippocampo accompagnata da deficit cognitivi significativi, aumento della fosforilazione di TAU e depositi di A-42 [54].
NRF2 Up-Regola i geni chiave per il mantenimento della Fisiologia dell'ER
Il lume ER necessita di un'abbondante fornitura di GSH dal citosol per mantenere la chimica del disolfuro. NRF2 modula enzimi cruciali del metabolismo GSH nel cervello, come il trasporto di cistina / glutammato,? -Glutammato cisteina sintetasi (? -GS), glutammato-cisteina ligasi catalitica e subunità modulatrici (GCLC e GCLM), glutatione reduttasi (GR) e glutatione perossidasi (GPX) (rivisto in [55]). La rilevanza di NRF2 nel mantenimento di GSH in ER è supportata dalla scoperta che l'attivazione farmacologica o genetica di NRF2 si traduce in una maggiore sintesi di GSH tramite GCLC / GCLM, mentre l'inibizione dell'espressione di questi enzimi da NRF2-knockdown ha causato un accumulo di danni proteine all'interno del pronto soccorso che portano all'attivazione di UPR [56].
In C. elegans diversi componenti dei geni bersaglio UPR regolati da SKN-1, inclusi Ire1, Xbp1 e Atf6. Sebbene NRF2 regoli l'espressione di diversi geni della perossidasi (PRX) e della glutatione perossidasi (GPX) nei mammiferi (rivisto in [57]), solo GPX8 è un enzima ER localizzato in buona fede, che ospita il segnale di recupero KDEL [58]. La perdita di GPX8 causa l'attivazione di UPR, la fuoriuscita di perossido di idrogeno derivato da ERO1? Nel citosol e la morte cellulare. Perossido di idrogeno derivato da ERO1? l'attività non può diffondersi dal pronto soccorso al citosol a causa dell'azione concertata di GPX8 e PRX4 [59]. A questo proposito, un'analisi della matrice di espressione genica del percorso di difesa antiossidante utilizzando RNA da tessuto di topo wild type e NRF2-null, ha rivelato che l'espressione di GPX8 era down-regolata in assenza di NRF2 [60]. In linea con ciò, l'analisi del trascrittoma da campioni di pazienti affetti da neoplasie mieloproliferative, policitemia o mielofibrosi, malattie associate anche a stress ossidativo e infiammazione cronica di basso grado, mostrano livelli di espressione più bassi sia di NRF2 che di GPX8 rispetto ai soggetti di controllo [61]. Non ci sono ancora studi che coinvolgono specificamente la GPX8 nella protezione del cervello umano, ma un'analisi del trascrittoma nei topi indica un aumento compensatorio della GPX8 in risposta alla tossina parkinsoniana MPTP [62].
Impatto di NRF2 sulla disregolazione UPR nelle malattie neurodegenerative
Il malfunzionamento degli enzimi PDI e l'attivazione cronica dell'UPR potrebbero successivamente avviare o accelerare la neurodegenerazione. Neuroni colpiti dalla malattia, modelli animali di malattie neurodegenerative e tessuti umani post-mortem hanno evidenziato l'up-regulation di diversi marcatori UPR nella maggior parte di questi disturbi. L'alterazione del pathway PDI / UPR nelle malattie neurodegenerative è stata ben rivista in [63] ma i seguenti punti salienti dei campioni post-mortem del cervello dovrebbero essere considerati. I livelli di PDI sono aumentati nei neuroni portatori di tanghi e nei corpi di Lewy dei pazienti con AD e PD, rispettivamente [64], [65]. PDI ed ERP57 sono regolati in alto nel CSF da pazienti con SLA e nel cervello da soggetti con CJD [66], [67], [68]. BIP, PERK, IRE1 e ATF6 sono elevati nei campioni da pazienti con AD, PD o ALS [69], [70], [71], [67]. BIP, CHOP e XBP1 sono elevati nei campioni cerebrali post-mortem da HD [72], [73]. Inoltre, l'up-regulation di ERP57, GRP94 e BIP è stato trovato nei tessuti della corteccia da pazienti con CJD [74]. Complessivamente, questa evidenza rivela che l'accumulo di proteine mal ripiegate nel parenchima cerebrale porta a un'attivazione deleterio e cronica dell'UPR. È interessante notare che c'è uno studio recente che collega l'attivazione di NRF2 con il PERK all'inizio dell'AD. In questo studio, gli autori hanno analizzato se i cambiamenti mediati dallo stress ossidativo in NRF2 e UPR possono costituire eventi precoci nella patogenesi dell'AD utilizzando le cellule del sangue periferico umano e un modello di topo transgenico AD in diverse fasi della malattia. Aumento dello stress ossidativo e aumento di pSer40-NRF2 sono stati osservati nelle cellule mononucleate del sangue periferico umano isolate da individui con compromissione cognitiva lieve. Inoltre, hanno segnalato una compromissione dell'omeostasi del calcio ER e dei marcatori di stress ER sovrastimolati in queste cellule da individui con compromissione cognitiva lieve e lieve AD [75].
Mutua regolamentazione di NRF2 e Ubiquitin Proteasome System (UPS)
L'UPS modula i livelli di proteine NRF2
L'UPS partecipa alla degradazione delle proteine danneggiate o malriposte e controlla i livelli delle molecole regolatrici chiave nel citosol e nel nucleo. Il nucleo centrale di questo sistema è un grande enzima multisubunit che contiene un complesso proteolitico attivo denominato 20S. Il proteasoma core 20S degrada le proteine dispiegate, ma il legame con i diversi complessi proteici regolatori ne modifica la specificità e l'attività del substrato. Ad esempio, l'aggiunta di una o due subunità regolatorie 19S al core 20S costituisce il proteasoma 26S e cambia la sua specificità rispetto alle proteine native piegate [76], [77]. La degradazione del proteasomal ha bisogno del legame covalente dell'ubiquitina. La coniugazione dell'ubiquitina procede attraverso un meccanismo a cascata in tre fasi. Innanzitutto, l'enzima che attiva l'ubiquitina E1 attiva l'ubiquitina in una reazione che richiede ATP. Quindi, un enzima E2 (ubiquitina-carrier protein o ubiquitin-conjugating enzyme) trasferisce l'ubiquitina attivata da E1 al substrato che è specificamente legato a un membro della famiglia delle ligasi della proteina ubiquitina, chiamata E3. Sebbene il destino esatto della proteina ubiquitinata dipenda dalla natura della catena dell'ubiquitina, questo processo generalmente provoca la degradazione del proteasoma 26S [78].
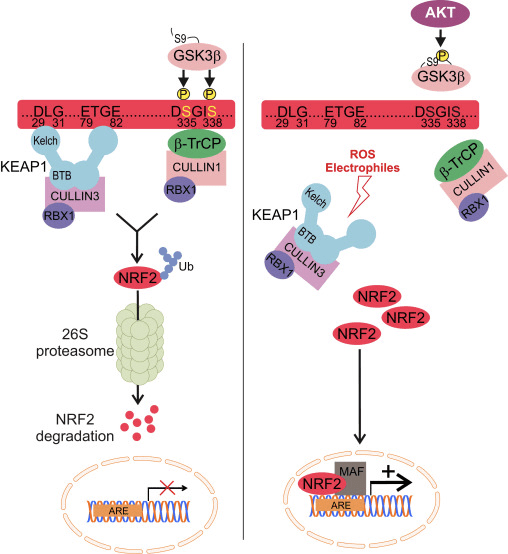
L'E3-ligasi KEAP1 è il più noto inibitore di NRF2. Il meccanismo del regolamento KEAP1 spiega elegantemente come i livelli di NRF2 si adattano alle fluttuazioni ossidanti. In condizioni basali, NRF2 recentemente sintetizzato viene afferrato dall'omodimero KEAP1, che lega una molecola NRF2 a due sequenze di ammino acidi con affinità bassa (aspartato, leucina, glicina; DLG) e alta (glutammato, treonina, glicina, glutammato, ETGE). L'interazione con KEAP1 aiuta a presentare NRF2 al complesso proteico CULLIN3 / RBX1, determinando la sua ubiquitinazione e la conseguente degradazione del proteasomal. Tuttavia, la modifica redox di KEAP1 impedisce la presentazione di NRF2 all'UPS rappresentato da CULLIN3 / RBX1. Di conseguenza, NRF2 appena sintetizzato sfugge alla degradazione dipendente da KEAP1, si accumula nel nucleo e attiva i geni contenenti ARE [79], [80], [81], [82].
L'adattatore E3-ligasi? -TrCP è anche un omodimero che partecipa agli eventi di segnalazione relativi alla fosforilazione di NRF2 da parte di GSK-3 ?. Questa chinasi fosforila residui di serina specifici di NRF2 (aspartato, serina, glicina, isoleucina serina; DSGIS) per creare un dominio di degradazione che viene quindi riconosciuto da? -TrCP e contrassegnato per la degradazione del proteasoma da un complesso CULLIN1 / RBX1. L'identificazione degli amminoacidi specifici che sono fosforilati da GSK-3? in questo degron è stata condotta una combinazione di mutagenesi sito-diretta del dominio Neh6, elettroforesi su gel 2D [15], [26] e spettroscopia di massa [83]. Di conseguenza, l'inibizione di GSK-3? da farmaci altamente selettivi o siRNA contro le isoforme GSK-3 ha determinato un aumento dei livelli di proteina NRF2. Risultati simili sono stati trovati con siRNA contro le isoforme di? -TrCP 1 e 2. Stabilizzazione di NRF2 dopo GSK-3? l'inibizione si è verificata in fibroblasti embrionali di topo KEAP1-carenti e in un mutante di delezione NRF2 espresso ectopicamente privo dei residui ETGE critici per il legame ad alta affinità con KEAP1, dimostrando ulteriormente una regolazione indipendente da KEAP1.
Nel contesto delle malattie neurodegenerative, possiamo prevedere la modulazione di NRF2 da parte dell'UPS in due modi diversi. Da un lato, il sistema KEAP1 rileverebbe uno squilibrio redox derivato dall'accumulo di proteine mal ripiegate, mentre l'asse GSK-3 /? - TrCP agirebbe come un partecipante attivo nella trasduzione del segnale alterata dalla perdita di proteostasi (Fig.2).
NRF2 aumenta l'attività dell'UPS attraverso il controllo trascrizionale delle subunità proteasiche
NRF2 regola l'espressione di diverse subunità del proteosoma, proteggendo così la cellula dall'accumulo di proteine tossiche. Venti geni correlati al proteasoma e all'ubiquitinazione sembrano essere regolati da NRF2, secondo un'ampia analisi di microarray da RNA epatico che è stata impostata con l'induttore NRF2 D3T [84]. In uno studio posteriore, gli stessi autori hanno evidenziato che l'espressione della maggior parte delle subunità del proteasoma 26S era aumentata fino a tre volte nel fegato di topi trattati con D3T. I livelli di proteine della subunità e l'attività del proteasoma sono stati aumentati in modo coordinato. Tuttavia, nessuna induzione è stata osservata nei topi in cui il fattore di trascrizione NRF2 è stato interrotto. L'attività del promotore della subunità proteasoma PSMB5 (20S) è aumentata con la sovraespressione di NRF2 o il trattamento con attivatori nei fibroblasti embrionali di topo e le ARE sono state identificate nel promotore prossimale di PSMB5 [85]. L'attivazione farmacologica di NRF2 ha portato a livelli di espressione elevati di subunità rappresentative del proteasoma (PSMA3, PSMA6, PSMB1 e PSMB5) solo in fibroblasti umani non senescenti contenenti NRF2 funzionale [86]. L'attivazione di NRF2 durante l'adattamento allo stress ossidativo determina un'elevata espressione di PSMB1 (20S) e PA28? subunità (o S11, regolatore del proteasoma) [87]. Inoltre, i risultati delle cellule staminali embrionali umane hanno rivelato che NRF2 controlla l'espressione della proteina di maturazione del proteasoma (POMP), un chaperone del proteasoma, che a sua volta modula la proliferazione di cellule staminali embrionali umane che si auto-rinnovano, la differenziazione dei tre strati germinali e la riprogrammazione cellulare [ 88]. Tutti insieme, questi studi indicano che NRF2 regola l'espressione dei componenti chiave dell'UPS e quindi contribuisce attivamente alla clearance delle proteine che altrimenti sarebbero tossiche.
L'asse NRF2-UPS nelle malattie neurodegenerative
Il ruolo dell'UPS nelle malattie neurodegenerative è un campo di intenso dibattito. Gli studi iniziali hanno riportato una diminuzione dell'attività proteasoma nelle necroscopie umane di pazienti affetti da diverse malattie neurodegenerative. Tuttavia, altri studi che impiegano approcci in vitro e in vivo hanno rilevato un'attività proteasoma invariata o addirittura aumentata (revisionata in [89]). Una possibile spiegazione di questa discrepanza è che i livelli dei componenti dell'UPS potrebbero cambiare durante la progressione della malattia e in diverse regioni del cervello, come è stato suggerito per gli obiettivi NRF2.
Nonostante questa controversia, va notato che l'up-regulation dei geni del proteasoma contenenti ARE rafforzerà l'UPS aumentando la clearance delle proteine tossiche nel cervello. Infatti, l'ablazione di NRF1, anche modulatore della risposta antiossidante, nelle cellule neuronali porta a compromissione dell'attività del proteasoma e della neurodegenerazione. Gli esperimenti di immunoprecipitazione della cromatina e l'analisi trascrizionale hanno dimostrato che PSMB6 è regolato da NRF1. Inoltre, il profilo di espressione genica ha portato all'identificazione di NRF1 come regolatore chiave trascrizionale dei geni del proteasoma nei neuroni, suggerendo che le perturbazioni in NRF1 possono contribuire alla patogenesi delle malattie neurodegenerative [90]. È interessante notare che NRF1 e la sua isoforma lunga denominata TCF11 hanno mostrato di up-regolare i geni del proteasoma contenenti ARE sull'inibizione del proteasoma in un anello di feedback per compensare la ridotta attività proteolitica [91], [92].
Per quanto riguarda NRF2, esiste una correlazione tra la riduzione dei livelli di NRF2, RPT6 (19 S) e PSMB5 (20 S) nel mesencefalo di topi con deficit di DJ-1 trattati con il paraquat della neurotossina [93]. Inoltre, il composto sulforafano (SFN) presente in natura fornisce un'immagine più robusta di NRF2 come un modulatore cruciale dell'UPS. Esperimenti in vitro con neuroblastoma murino Le cellule di Neuro2A hanno evidenziato una maggiore espressione delle subunità catalitiche del proteasoma, nonché le sue attività peptidasi in risposta a SFN. Questo farmaco ha protetto le cellule dalla citotossicità perossido di idrogeno e dall'ossidazione delle proteine in modo dipendente dalla funzione del proteasoma [94]. Inoltre, Liu e collaboratori hanno impiegato un topo reporter per monitorare l'attività dell'UPS in risposta a SFN nel cervello. Questi topi esprimono ubiquitariamente la proteina di fluorescenza verde (GFP) fusa con un segnale di degradazione costitutiva che promuove la sua rapida degradazione da parte dell'UPS (GFPu). Nella corteccia cerebrale, SFN ha ridotto il livello di GFPu con un aumento parallelo delle attività simili a chimotripsina (PSMB5), caspase (PSMB2) e trypsin-like (PSMB1) del proteasoma 20 S. Inoltre, il trattamento delle cellule derivate da Huntington con SFN ha rivelato che l'attivazione di NRF2 ha migliorato la degradazione di mHtt e ridotto la citotossicità di mHtt [95]. Il principale meccanismo dell'azione SFN è attraverso l'induzione di NRF2 [96]. Il contributo specifico di NRF2 deve essere affrontato impiegando sistemi NRF2-null in ulteriori studi.
Connessione funzionale tra NRF2 e Macroautophagy
I livelli delle proteine NRF2 sono modulati dalla proteina dell'adattatore P62
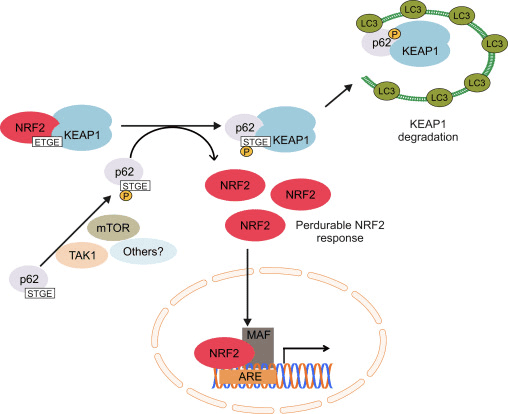
L'autofagia si riferisce alla degradazione dei componenti citosolici all'interno dei lisosomi. Questo processo viene utilizzato per la rimozione di proteine a lunga vita e malriposte e di organelli danneggiati. Un collegamento diretto tra NRF2 e autofagia è stato osservato per la prima volta in relazione alla proteina dell'adattatore p62, anche denominata SQSTM1 [97], [98], [99], [100], [101]. Questa proteina trasferisce le proteine ubiquitinate alle macchine di degradazione proteasomale e lisosomiale e sequestra le proteine danneggiate in aggregati prima della loro degradazione. P62 presenta un dominio UBA (ubiquitin-associated), per il legame alle proteine ubiquitinate e una regione LIR (LC3-interacting) per l'integrazione con la membrana autofagosomica attraverso il recettore dell'autofagia LC3.
Sebbene l'induzione mediata da p62 di NRF2 e dei suoi geni bersaglio sia stata segnalata per la prima volta nel 2007 [102], il meccanismo molecolare non è stato completamente compreso fino alla scoperta della sua interazione con KEAP1 [103], [98], [99], [100 ], [101]. Komatsu e colleghi hanno identificato una regione di interazione KEAP1 (KIR) in p62 che legava KEAP1 nella stessa tasca superficiale di base di NRF2 e con un'affinità di legame simile al motivo ETGE in NRF2, suggerendo una competizione tra p62 e NRF2. La fosforilazione di Ser351 nel motivo KIR in p62 (349-DPSTGE-354) ha dimostrato di aumentare la sua affinità per KEAP1, competendo con il legame NRF2 e consentendo il suo accumulo e l'attivazione trascrizionale dei suoi geni bersaglio [98], [99]. Infatti, la sovraespressione di p62 ha portato alla riduzione dell'ubiquitinazione di NRF2 e alla conseguente stabilizzazione, nonché all'induzione dei suoi geni bersaglio [104]. È stato suggerito che alcune chinasi partecipino alla fosforilazione di p62. Il bersaglio dei mammiferi del complesso rapamicina 1 (mTORC1) può essere implicato, poiché il trattamento con l'inibitore di mTOR rapamicina ha soppresso la fosforilazione di p62 e la sottoregolazione di KEAP1 dopo il trattamento con arsenite. Recentemente, è stato dimostrato che TGF -? - chinasi 1 attivata (TAK1) potrebbe anche fosforilare p62, migliorando la degradazione di KEAP1 e la regolazione di NRF2. Gli autori di questo studio suggeriscono che questo sia un modo per regolare la redoxtasis cellulare in condizioni di stato stazionario, poiché la carenza di TAK1 aumenta la regolazione dei ROS in assenza di qualsiasi ossidante esogeno in diversi tessuti di topo in parallelo con una riduzione dei livelli di proteina NRF2 [105 ].
Un costrutto p62 privo del dominio UBA era ancora in grado di legare KEAP1, implicando che l'interazione non dipendesse da KEAP1 ubiquitinato [101]. Tuttavia, l'omologo di p62 in Drosophila melanogaster, denominato Ref (2), non contiene un motivo KIR e non interagisce direttamente con DmKEAP1, sebbene possa legarsi a DmKEAP1 ubiquitinato attraverso il dominio UBA. Inoltre, DmKEAP1 può interagire direttamente con Atg8 (omologo ai mammiferi LC3). Il deficit di KEAP1 risulta in Atg8 e l'induzione dell'autofagia dipendente dall'ortologo di NRF2 CncC e indipendente da TFEB / MITF [106]. La relazione tra NRF2 e autofagia sembra essere conservata, evidenziando la sua rilevanza funzionale.
L'induzione di NRF2 da parte di p62 è il risultato di entrambe le gare per legare KEAP1 e la degradazione di KEAP1 nel lisosoma. Il silenziamento di p62 con siRNA ha raddoppiato l'emivita di KEAP1 in parallelo con una diminuzione di NRF2 e dei suoi geni bersaglio [101]. In accordo, l'ablazione dell'espressione di p62 ha evidenziato un aumento dei livelli di KEAP1 rispetto ai topi wild type. Molto rilevante, l'incremento dei livelli di KEAP1 non è stato influenzato dagli inibitori del proteasoma, ma è stato ridotto con l'autophagy inducente la fame [107]. Infatti, KEAP1 è presente nelle cellule di mammifero in vescicole autofagiche decorate con p62 e LC3 [99], [100], [103]. Tutti questi dati suggeriscono che KEAP1 è un substrato del macchinario di macroautofagia, ma questo problema dovrebbe essere analizzato con maggiori dettagli a causa dell'esistenza di alcuni risultati controversi. I livelli di proteina KEAP1 sono aumentati nei topi Atg7 null, un effettore chiave della macroautofagia [107], ma l'inibizione farmacologica della macroautofagia con torin1, E64 / pepstatin o bafilomicina non ha accumulato KEAP1 [107], [100]. Nel complesso, questi risultati suggeriscono che l'aumento dei livelli di p62 sequestra KEAP1 in vacuoli autofagici e probabilmente questi risultati nella degradazione autofagica di KEAP1 permettendo l'attivazione di NRF2 (Fig. 3). Due diversi studi hanno riportato che l'acido sulfinico riduttasi SESTRINS svolge un ruolo importante in questo contesto. SESTRIN 2 interagisce con p62, KEAP1 e RBX1 e facilita la degradazione dipendente da p62 dell'attivazione di KEAP1 e NRF2 dei geni target [108]. Un altro studio ha dimostrato che SESTRIN 2 interagiva con ULK1 e p62, promuovendo la fosforilazione di p62 a Ser403 che ha facilitato il degrado delle proteine del carico incluso KEAP1 [109].
Modulazione dei geni Macroautophagy di NRF2
NRF2 regola l'espressione di geni rilevanti per la macroautofagia, così come per l'UPR e l'UPS. La prima evidenza proviene da studi in cui è stato dimostrato che l'espressione di p62 è indotta dall'esposizione a elettrofili, ROS e ossido nitrico [110], [111], [112]. Il meccanismo di induzione è stato descritto alcuni anni dopo con la scoperta che p62 contiene un ARE funzionale nel suo promotore genico [99]. In un recente studio, sono state trovate diverse altre ARE funzionali e validate in seguito a analisi bioinformatica e analisi ChIP. Inoltre, i fibroblasti embrionali di topo e i neuroni corticali dei topi knockout di Nrf2 presentavano un'espressione ridotta di p62, che poteva essere salvata con un lentivirus che esprimeva NRF2. Allo stesso modo, il deficit di NRF2 ha ridotto i livelli di p62 nei neuroni feriti da topi ippocampo [36]. Pertanto, è stato suggerito che l'attivazione di NRF2 aumenta i livelli di p62, con conseguente degradazione di KEAP1 e favorendo l'ulteriore stabilizzazione di NRF2 in un ciclo di feedback positivo. Questo meccanismo non canonico di induzione NRF2 richiede cambiamenti nell'espressione genica e potrebbe essere una risposta rilevante allo stress cellulare prolungato.
La proteina di riconoscimento del carico NDP52 ha dimostrato di essere regolata per via trascrizionale da NRF2. NDP52 funziona in modo simile a p62, riconoscendo proteine ubiquitinate e interagendo con LC3 attraverso un dominio LIR, in modo che i carichi siano degradati nei lisosomi. Sono stati trovati cinque ipotetici valori nella sequenza del DNA del promotore Ndp52. Tre di essi sono stati identificati con diversi costrutti mutanti e test ChIP come indispensabili per la trascrizione Ndp2 mediata da NRF52 [113]. Di nota, i livelli di mRNA di Ndp52 sono stati ridotti nell'ippocampo di topi knockout Nrf2. Una di queste sequenze è stata anche validata in uno studio indipendente come ARE [2] regolato da NRF36.
Tuttavia, il ruolo di NRF2 nella modulazione dell'autofagia non è limitato all'induzione di queste due proteine di riconoscimento del carico. Al fine di ottenere una visione più approfondita del ruolo di NRF2 nella modulazione di ulteriori geni correlati all'autofagia, il nostro gruppo ha esaminato il database di immunoprecipitazione della cromatina ENCODE per due proteine, MAFK e BACH1, che legano le ARE regolate da NRF2. Usando uno script generato dalla sequenza ARE di consenso di JASPAR, abbiamo identificato diverse POS putative in molti geni dell'autofagia. Dodici di queste sequenze sono state convalidate come ARE regolate da NRF2 in nove geni autofagici, la cui espressione è stata ridotta nei fibroblasti di embrioni di topo di topi knockout Nrf2, ma potrebbe essere ripristinata da un lentivirus che esprime NRF2. Il nostro studio ha dimostrato che NRF2 attiva l'espressione di alcuni geni coinvolti in diverse fasi del processo autofagico, tra cui l'inizio dell'autofagia (ULK1), il riconoscimento del carico (p62 e NDP52), la formazione di autofagosomi (ATG4D, ATG7 e GABARAPL1), l'allungamento (ATG2B e ATG5) ) e clearance del autososoma (ATG4D). Di conseguenza, il flusso di autofagia in risposta al perossido di idrogeno è stato compromesso quando NRF2 era assente [36].
Rilevanza Dell'espressione Genica Dei Macroautofagia Mediata Da NRF2 Nei Disturbi Neurodegenerativi
L'autofagia difettosa ha dimostrato di svolgere un ruolo importante in diverse malattie neurodegenerative [114] e l'ablazione dell'autofagia porta alla neurodegenerazione nei topi [115], [116]. Topi knockout di Atg7 hanno rivelato che il deficit di autofagia si traduce in un accumulo di p62 in corpi di inclusione ubiquitina-positivi. KEAP1 è stato sequestrato in questi corpi di inclusione, portando alla stabilizzazione di NRF2 e all'induzione dei geni bersaglio [103]. È importante notare che l'accumulo eccessivo di p62 insieme a proteine ubiquitinate è stato identificato nelle malattie neurodegenerative, tra cui AD, PD e ALS [117]. Infatti, i neuroni che esprimono alti livelli di APP o TAU di pazienti con AD hanno espresso anche p62 e NRF2 nucleare, suggerendo il loro tentativo di degradare gli aggregati intraneuronali attraverso l'autofagia [36].
La carenza di NRF2 aggrava l'aggregazione proteica nel contesto dell'AD. Infatti, livelli aumentati di TAU fosforilata e insolubile in sarkosil si trovano nei topi knockout per Nrf2, sebbene non sia stata rilevata alcuna differenza nelle attività della chinasi o della fosfatasi confrontandola con lo sfondo wild-type [113]. È importante sottolineare che NDP52 è stato dimostrato di co-localizzare con TAU nei neuroni murini e l'interazione diretta tra fosfo-TAU e NDP52 è stata dimostrata da esperimenti di co-immunoprecipitazione sia nei topi che nei campioni di AD, indicando il suo ruolo nella degradazione della TAU. È interessante notare che il silenziamento di NDP52, p62 o NRF2 nei neuroni ha portato a un aumento della fosfo-TAU [113], [118]. Inoltre, sono stati trovati aggregati APP intraneuronali aumentati nell'ippocampo di topi APP / PS1? E9 quando NRF2 era assente. Ciò era correlato a marcatori di autofagia alterati, tra cui un aumento dei rapporti fosfo-mTOR / mTOR e fosfo-p70S6k / p70S6k (indicativi di inibizione dell'autofagia), livelli aumentati di pre-catepsina D e un numero maggiore di corpi multivesicolari [119]. Nei topi che coesprimono APP umana (V717I) e TAU (P301L), il deficit di NRF2 ha portato ad un aumento dei livelli di TAU totale e fosfo-TAU nella frazione insolubile e ad un aumento degli aggregati di APP intraneuronali, insieme a livelli neuronali ridotti di p62, NDP52, ULK1, ATG5 e GABARAPL1. La co-localizzazione tra la proteina adattatore p62 e APP o TAU è stata ridotta in assenza di NRF2 [36]. Nel complesso, questi risultati evidenziano l'importanza di NRF2 nell'autofagia neuronale.
I diversi fattori di trascrizione agiscono coordinatamente per modulare la proteostasi
In condizioni di stato stazionario, la proteostasi viene controllata tramite interazioni proteina-proteina e modificazioni post-traduzionali ottenendo una risposta rapida. Tuttavia, l'adattamento cellulare richiede la regolazione trascrizionale dei geni UPR, UPS e autofagia. Considerando che le cellule nervose sono continuamente sottoposte a insulti tossici di basso grado, tra cui lo stress ossidativo e proteotossico, un rinforzo della proteostasi indotta dalla modulazione trascrizionale potrebbe aiutare a prevenire la degenerazione del cervello.
Nel caso dell'UPR, l'attivazione di ciascuna delle tre braccia porterà infine all'induzione trascrizionale di alcuni geni (rivisti in [43]). Ad esempio, un frammento derivato da ATF6 (ATF6f) si lega agli elementi di risposta allo stress ER (ERSE) e induce l'espressione di diversi geni, tra cui XBPI, BIP e CHOP. Inoltre, la segnalazione PERK porta all'attivazione del fattore di trascrizione ATF4, che controlla l'espressione di più geni correlati a UPR e alcuni altri tra cui i geni bersaglio NRF2 Hmox1 e p62. Infine, l'attivazione di IRE1 determina la generazione di un fattore di trascrizione attivo XBP1 (XBP1s), che controlla la trascrizione dei geni che codificano le proteine coinvolte nel ripiegamento delle proteine.
D'altra parte, NRF1 si è dimostrato necessario per l'espressione genica del proteasomal nel cervello, in quanto i topi knockout Nrf1 hanno esibito un'espressione ridotta di geni che codificano varie subunità del core 20S, così come il complesso regolatore 19S insieme a funzionalità proteasomica compromessa [90 ]. Sia NRF1 che NRF2 si legano alle sequenze ARE nelle regioni del promotore dei suoi geni bersaglio, il che suggerisce che hanno attività trascrizionali sovrapposte, sebbene differiscano nei loro meccanismi regolatori e nella localizzazione cellulare [120].
I fattori di trascrizione della famiglia Forkhead box O (FOXO) controllano l'espressione di più geni correlati all'autofagia. Simile a ciò che avviene con NRF2, ci sono più livelli di regolazione dell'attività dei membri FOXO, che possono essere indotti su stress nutrizionale o ossidativo [121]. Infine, il fattore di trascrizione TFEB, considerato il principale regolatore della biogenesi lisosomiale, svolge un ruolo cruciale nella regolazione dell'autofagia in condizioni di stress nutrizionale. Pertanto, l'inibizione di mTORC1 porta alla traslocazione nucleare di TFEB e all'induzione dell'espressione di geni autofagici [122].
Nel complesso, l'esistenza di diversi regolatori trascrizionali di questi macchinari suggerisce anche diafonia e meccanismi parzialmente ridondanti che possono assicurare la proteostasi in diverse circostanze. Di conseguenza, NRF2 può avere un ruolo rilevante nei tessuti che supportano alti livelli di stress ossidativo. Ad esempio, NRF2 indotto da stress ossidativo può funzionare in condizioni ricche di sostanze nutritive per autophagy sovraregolare in modo trascrizionale, simile a quanto è stato trovato per TFEB in condizioni di fame. Inoltre, il cervello funziona in gran parte sotto condizioni nutritive, ponendo NRF2 come un meccanismo rilevante per attivare l'autofagia nei neuroni.
Promettente Potenziale Terapeutico per NRF2 nelle Proteinopatie
Negli ultimi anni sono stati compiuti notevoli progressi nella conoscenza dei ruoli normativi dell'UPR, dell'UPS e dell'autofagia sull'attività di NRF2, nonché della trascrizione reciproca mediata da NRF2 di componenti di questi tre sistemi. Pertanto, possono sorgere nuove possibilità terapeutiche basate sullo sfruttamento di NRF2 come regolatore cruciale della clearance delle proteine nelle malattie neurodegenerative.
Tuttavia, una questione chiave che rimane è se sarà utile o deleterio aumentare i livelli di NRF2 nel cervello. L'analisi dei dati epidemiologici può fornire una risposta parziale, in quanto indica che il gene NFE2L2 è altamente polimorfico e alcuni polimorfismi a singolo nucleotide trovati nella sua regione regolatrice del promotore possono fornire una gamma di variabilità `` fisiologica '' nell'espressione genica a livello di popolazione e alcuni aplotipi erano associati a una riduzione del rischio e / o all'insorgenza ritardata di AD, PD o SLA [123]. Inoltre, come discusso da Hayes e colleghi [124], l'effetto NRF2 potrebbe avere una risposta a forma di U, il che significa che livelli troppo bassi di NRF2 possono provocare una perdita di citoprotezione e una maggiore suscettibilità ai fattori di stress, mentre una quantità eccessiva di NRF2 potrebbe disturbare l'equilibrio omeostatico verso uno scenario riduttivo (stress riduttivo), che favorirebbe il misfolding e l'aggregazione delle proteine. Bassi livelli di NRF2 nel cervello supportano l'idea che una leggera sovraregolazione possa essere sufficiente per ottenere un beneficio in condizioni patologiche. Infatti, il ruolo protettivo dell'attivazione farmacologica della clearance della proteina mediata da NRF2 è stato dimostrato in diverse colture cellulari di neurodegenerazione e modelli in vivo.
SFN è un attivatore farmacologico di NRF2 che ha dimostrato di indurre l'espressione genica del proteasoma e dell'autofagia [95], [36]. È interessante notare che Jo e colleghi hanno dimostrato che SFN riduce i livelli di TAU fosforilata e aumenta Beclin-1 e LC3-II, suggerendo che l'attivazione di NRF2 può facilitare la degradazione di questa proteina tossica attraverso l'autofagia [113]. Inoltre, la degradazione di mHtt è stata migliorata con SFN, e questo è stato invertito con l'uso di MG132, indicando la degradazione proteasomica di questa proteina tossica [95]. La degradazione mediata dall'autofagia di TAU fosfo e insolubile è stata segnalata con la fisetina flavonoide organica. Questo composto è stato in grado di indurre l'autofagia promuovendo simultaneamente l'attivazione e la traslocazione nucleare di TFEB e NRF2, insieme ad alcuni dei suoi geni bersaglio. Questa risposta è stata prevenuta dal silenziamento TFEB o NRF2 [125]. Bott e colleghi hanno riportato effetti benefici di un attivatore simultaneo di NRF2, NRF1 e HSF1 sulla tossicità delle proteine nell'atrofia muscolare spinale e bulbare, una malattia neurodegenerativa causata dall'espansione delle ripetizioni CAG codificanti la poliglutamina in cui sono presenti aggregati proteici [126]. Il potenziale di attivazione di NRF2 per il trattamento dei disturbi neurodegenerativi è stato dimostrato con l'approvazione di BG-12, la formulazione orale dell'induttore NRF2 dimetilfumarato (DMF), per il trattamento della sclerosi multipla [127], [128]. Il successo del DMF con malattie autoimmuni con una forte componente infiammatoria suggerisce che le malattie neurodegenerative potrebbero trarre beneficio dal riposizionamento di questo farmaco. In un recente studio preclinico su un modello di? -Sinucleinopatia di PD, la DMF ha dimostrato di essere neuroprotettiva a causa, in parte, della sua induzione di autofagia [129]. Gli studi che riportano gli effetti benefici di NRF2 sulla neurodegenerazione ma non si concentrano sul suo effetto sulla clearance delle proteine sono ancora più abbondanti (per una revisione completa, vedere [7]). Questo è abbastanza rilevante, in quanto evidenzia i molteplici processi dannosi che possono essere simultaneamente presi di mira da un singolo colpo in NRF2, inclusi anche lo stress ossidativo, la neuroinfiammazione o la disfunzione mitocondriale. Tuttavia, sarà necessario un lavoro futuro per determinare definitivamente se l'attivazione farmacologica di NRF2 può essere una strategia valida per facilitare la degradazione delle proteine tossiche nel cervello.
Come spiegato prima, GSK-3 esacerbato? l'attività è stata riportata nelle malattie neurodegenerative ed è stato ipotizzato che la conseguente riduzione di NRF2 possa essere parzialmente responsabile dell'outcome deleterio. In queste condizioni patologiche, gli inibitori GSK-3 potrebbero anche cooperare per aumentare i livelli di NRF2 e la proteostasi. Gli effetti benefici degli inibitori GSK-3 sono stati riportati in diversi modelli di neurodegenerazione e, cosa più interessante, è stato dimostrato che la repressione GSK-3 riduce i livelli di proteine tossiche [130], [131], [132], [133]. Sebbene non siano stati ancora osservati collegamenti diretti tra l'inibizione di GSK-3 e la regolazione trascrizionale di NRF2 dei geni che promuovono la proteostasi, è ragionevole ipotizzare che la sottoregolazione dell'attività GSK-3 comporterebbe un aumento dei livelli di NRF2, che alla fine si tradurrà in un rafforzamento proteostasi.
L'attività trascrizionale di NRF2 e la capacità cellulare di mantenere la proteostasi diminuiscono con l'età, principale fattore di rischio per lo sviluppo di malattie neurodegenerative. È ragionevole pensare che il rafforzamento di NRF2 e, di conseguenza, la proteostasi ritarderebbe almeno l'accumulo di aggregati proteici e la neurodegenerazione. Infatti, il trattamento dei fibroblasti senescenti umani con triterpenoide di acido 18? -Glicirretinico (18? -GA) ha promosso l'attivazione di NRF2, portando all'induzione del proteasoma e a una maggiore durata della vita. Questo studio suggerisce che l'attivazione farmacologica di NRF2 è possibile anche in tarda età [86]. Inoltre, uno studio successivo ha mostrato che questo composto mediava SKN-1 e l'attivazione del proteasoma in C.elegans con effetti benefici sulla progressione dell'AD in modelli di nematodi rilevanti [134].
Tutto sommato, l'induzione mediata da NRF2 di geni correlati alla proteostasi sembra essere benefica in diverse proteinopatie.
Sulforafano e suoi effetti su cancro, mortalità, invecchiamento, cervello e comportamento, malattie cardiache e altro
Gli isotiocianati sono alcuni dei composti vegetali più importanti che si possono ottenere nella dieta. In questo video faccio il caso più completo per loro che sia mai stato fatto. Soglia di attenzione breve? Passa al tuo argomento preferito facendo clic su uno dei seguenti punti temporali. Timeline completa di seguito.
Sezioni chiave:
- 00: 01: 14 - Cancro e mortalità
- 00: 19: 04 - Invecchiamento
- 00: 26: 30 - Cervello e comportamento
- 00: 38: 06 - Riassunto finale
- 00: 40: 27 - Dose
Timeline completa:
- 00: 00: 34 - Introduzione di sulforaphane, uno degli obiettivi principali del video.
- 00: 01: 14 - Consumo di verdure crocifere e riduzione della mortalità per tutte le cause.
- 00: 02: 12 - Rischio di cancro alla prostata.
- 00: 02: 23 - Rischio di cancro alla vescica.
- 00: 02: 34 - Carcinoma polmonare nei fumatori.
- 00: 02: 48 - Rischio di cancro al seno.
- 00: 03: 13 - Ipotetico: cosa succede se hai già un cancro? (Interventistica)
- 00: 03: 35 - Meccanismo plausibile che guida i dati associativi sul cancro e sulla mortalità.
- 00: 04: 38 - Sulforaphane e cancro.
- 00: 05: 32 - Prova animale che mostra un forte effetto dell'estratto di germogli di broccolo sullo sviluppo del tumore della vescica nei ratti.
- 00: 06: 06 - Effetto dell'integrazione diretta di sulforafano nei pazienti affetti da cancro alla prostata.
- 00: 07: 09 - Bioaccumulo di metaboliti di isotiocianato nel tessuto mammario effettivo.
- 00: 08: 32 - Inibizione delle cellule staminali del carcinoma mammario.
- 00: 08: 53 - Lezione di storia: le brassiche sono state istituite come aventi proprietà sanitarie anche nell'antica Roma.
- 00: 09: 16 - La capacità del Sulforaphane di potenziare l'escrezione di cancerogeno (benzene, acroleina).
- 00: 09: 51 - NRF2 come interruttore genetico tramite elementi di risposta antiossidante.
- 00: 10: 10 - Come l'attivazione di NRF2 migliora l'escrezione di cancerogeno tramite glutatione-S-coniugati.
- 00: 10: 34 - I cavoletti di Bruxelles aumentano la glutatione-S-transferasi e riducono il danno al DNA.
- 00: 11: 20 - La bevanda di germogli di broccoli aumenta l'escrezione di benzene di 61%.
- 00: 13: 31 - L'omogenato di germogli di broccoli aumenta gli enzimi antiossidanti nelle vie aeree superiori.
- 00: 15: 45 - Consumo di verdure crocifere e mortalità per malattie cardiache.
- 00: 16: 55 - La polvere di germogli di broccoli migliora i lipidi nel sangue e il rischio complessivo di malattie cardiache nei diabetici di tipo 2.
- 00: 19: 04 - Inizio della sezione di invecchiamento.
- 00: 19: 21 - La dieta arricchita con sulforafano migliora la durata della vita dei coleotteri da 15 a 30% (in determinate condizioni).
- 00: 20: 34 - L'importanza della bassa infiammazione per la longevità.
- 00: 22: 05 - Le verdure crocifere e la polvere di germogli di broccoli sembrano ridurre un'ampia varietà di marcatori infiammatori negli esseri umani.
- 00: 23: 40 - Ricapitolazione di metà video: cancro, sezioni di invecchiamento
- 00: 24: 14 - Gli studi sui topi suggeriscono che il sulforafano potrebbe migliorare la funzione immunitaria adattativa in età avanzata.
- 00: 25: 18 - Sulforaphane ha migliorato la crescita dei peli in un modello murino di calvizie. Immagine su 00: 26: 10.
- 00: 26: 30 - Inizio della sezione cervello e comportamento.
- 00: 27: 18 - Effetto dell'estratto di germogli di broccoli sull'autismo.
- 00: 27: 48 - Effetto del glucorapanin sulla schizofrenia.
- 00: 28: 17 - Inizio della discussione sulla depressione (meccanismo e studi plausibili).
- 00: 31: 21 - Lo studio del mouse utilizzando 10 diversi modelli di depressione indotta da stress mostra sulforapano in modo simile efficace come fluoxetina (prozac).
- 00: 32: 00 - Lo studio mostra che l'ingestione diretta di glucorafanina nei topi è altrettanto efficace nel prevenire la depressione dal modello di stress sociale di sconfitta.
- 00: 33: 01 - Inizio della sezione di neurodegenerazione.
- 00: 33: 30 - Sulforaphane e malattia di Alzheimer.
- 00: 33: 44 - Sulforaphane e morbo di Parkinson.
- 00: 33: 51 - Sulforaphane e la malattia di Hungtington.
- 00: 34: 13 - Sulforaphane aumenta le proteine da shock termico.
- 00: 34: 43 - Inizio della sezione traumatica di lesioni cerebrali.
- 00: 35: 01 - Sulforaphane iniettato immediatamente dopo TBI migliora la memoria (studio del mouse).
- 00: 35: 55 - Sulforaphane e plasticità neuronale.
- 00: 36: 32 - Sulforaphane migliora l'apprendimento nel modello di diabete di tipo II nei topi.
- 00: 37: 19 - Distrofia muscolare sulforapano e duchenne.
- 00: 37: 44 - Inibizione della miostatina nelle cellule muscolari satelliti (in vitro).
- 00: 38: 06 - Ricapitolazione tardiva: mortalità e cancro, danno al DNA, stress ossidativo e infiammazione, escrezione di benzene, malattie cardiovascolari, diabete di tipo II, effetti sul cervello (depressione, autismo, schizofrenia, neurodegenerazione), via NRF2.
- 00: 40: 27 - Pensieri sulla determinazione di una dose di germogli di broccoli o sulforafano.
- 00: 41: 01 - Aneddoti su germinazione a casa.
- 00: 43: 14 - Sulle temperature di cottura e sull'attività del sulforafano.
- 00: 43: 45 - Conversione batterica intestinale del sulforafano da glucorafanina.
- 00: 44: 24 - Gli integratori funzionano meglio se combinati con la mirosinasi attiva delle verdure.
- 00: 44: 56 - Tecniche di cottura e verdure crucifere.
- 00: 46: 06 - Isotiocianati come goitrogens.

Il fattore 2 (NF-E2), derivato da eritroide, derivato da eritroide, è un fattore di trascrizione che regola l'espressione di una varietà di enzimi antiossidanti e disintossicanti. Studi di ricerca hanno anche dimostrato il suo ruolo nel controllo dello stress ossidativo. La maggior parte delle malattie neurodegenerative, come il morbo di Alzheimer e il morbo di Parkinson, sono caratterizzate da stress ossidativo e infiammazione cronica, gli obiettivi comuni di Approcci trattamento Nrf2. Dr. Alex Jimenez DC, CCST Insight
Osservazioni conclusive
Il fattore di trascrizione NRF2 orchestra una risposta proteostatica rilevando e modulando i cambiamenti in UPR, UPS e autofagia (Fig. 4). Di conseguenza, la mancanza di NRF2 ha dimostrato di aggravare la proteinopatia, suggerendo che NRF2 è necessario per una clearance ottimale delle proteine. Tutti insieme, possiamo ipotizzare che NRF2 potrebbe essere un obiettivo terapeutico interessante per le proteinopatie.
Ringraziamenti
Sciencedirect.com/science/article/pii/S2213231716304050
Secondo l'articolo precedente, mentre i sintomi delle malattie neurodegenerative possono essere trattati attraverso una varietà di opzioni di trattamento, studi di ricerca hanno dimostrato che l'attivazione di Nrf2 può essere un approccio terapeutico utile. Perché Gli attivatori di Nrf2 prendono di mira ampi meccanismi di malattia, tutte le malattie neurodegenerative possono trarre vantaggio dall'uso del fattore di trascrizione Nrf2. I risultati di Nrf2 hanno rivoluzionato il trattamento delle malattie neurodegenerative. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
A cura di Dr. Alex Jimenez
Riferito da: Sciencedirect.com

Discussione aggiuntiva sull'argomento: alleviare il dolore al ginocchio senza chirurgia
Il dolore al ginocchio è un sintomo ben noto che può verificarsi a causa di una varietà di lesioni al ginocchio e / o condizioni, tra cui lesioni sportive. Il ginocchio è una delle articolazioni più complesse del corpo umano in quanto è costituito dall'intersezione di quattro ossa, quattro legamenti, vari tendini, due menischi e cartilagine. Secondo l'American Academy of Family Physicians, le cause più comuni di dolore al ginocchio includono sublussazione rotulea, tendinite rotulea o ginocchio del saltatore e malattia di Osgood-Schlatter. Sebbene il dolore al ginocchio sia più probabile che si verifichi nelle persone di età superiore ai 60 anni, il dolore al ginocchio può verificarsi anche nei bambini e negli adolescenti. Il dolore al ginocchio può essere trattato a casa seguendo i metodi RICE, tuttavia, gravi lesioni al ginocchio possono richiedere cure mediche immediate, compresa la cura chiropratica.

EXTRA EXTRA | ARGOMENTO IMPORTANTE: consigliato El Paso, TX Chiropractor
***
Le informazioni qui riportate su "Capire Nrf2 e il suo impatto sulle malattie neurodegenerative" non intende sostituire un rapporto individuale con un professionista sanitario qualificato o un medico autorizzato e non è una consulenza medica. Ti incoraggiamo a prendere decisioni sanitarie basate sulla tua ricerca e collaborazione con un professionista sanitario qualificato.
Informazioni sul blog e discussioni sull'ambito
Il nostro ambito informativo è limitato a chiropratica, muscolo-scheletrico, medicine fisiche, benessere, contributo eziologico disturbi viscerosomatici all'interno di presentazioni cliniche, dinamiche cliniche associate ai riflessi somatoviscerali, complessi di sublussazione, problemi di salute sensibili e/o articoli, argomenti e discussioni di medicina funzionale.
Forniamo e presentiamo collaborazione clinica con specialisti di varie discipline. Ogni specialista è regolato dal proprio ambito di pratica professionale e dalla propria giurisdizione di licenza. Utilizziamo protocolli funzionali di salute e benessere per trattare e supportare la cura delle lesioni o dei disturbi del sistema muscolo-scheletrico.
I nostri video, post, argomenti, soggetti e approfondimenti trattano questioni cliniche, problemi e argomenti che riguardano e supportano direttamente o indirettamente il nostro ambito di pratica clinica.*
Il nostro ufficio ha ragionevolmente tentato di fornire citazioni di supporto e ha identificato lo studio o gli studi di ricerca pertinenti a sostegno dei nostri post. Forniamo copie degli studi di ricerca di supporto a disposizione degli organi di regolamentazione e del pubblico su richiesta.
Comprendiamo che copriamo questioni che richiedono una spiegazione aggiuntiva su come può essere d'aiuto in un particolare piano di assistenza o protocollo di trattamento; pertanto, per discutere ulteriormente l'argomento di cui sopra, non esitate a chiedere Dott. Alex Jimenez, DC, o contattaci al 915-850-0900.
Siamo qui per aiutare te e la tua famiglia.
Blessings
Il dottor Alex Jimenez DC MSACP, Marina Militare*, CCST, IFMCP*, CIFM*, ATN*
e-mail: coach@elpasofunctionalmedicine.com
Licenza come Dottore in Chiropratica (DC) in Texas & Nuovo Messico*
Licenza Texas DC n. TX5807, Licenza DC del New Mexico n. NM-DC2182
Autorizzato come infermiere registrato (RN*) in Florida
Licenza Florida Licenza RN # RN9617241 (controllo n. 3558029)
Stato compatto: Licenza multistato: Autorizzato ad esercitare in Stati 40*
Dott. Alex Jimenez DC, MSACP, RN* CIFM*, IFMCP*, ATN*, CCST
Il mio biglietto da visita digitale





