L'eccitotossicità è caratterizzata come un insulto acuto che causa la morte delle cellule nervose a causa dell'eccessiva attivazione di iGluRs. L'eccitotossicità acuta gioca un ruolo fondamentale in una varietà di problemi di salute del sistema nervoso centrale (SNC), tra cui ischemia cerebrale, trauma cranico e stato epilettico. I meccanismi per l'eccitotossicità acuta sono diversi per ogni problema di salute.
Con l'ischemia cerebrale, l'eccitotossicità associata a L-glutammato e L-aspartato si verifica in pochi minuti a causa della crescita dell'L-glutammato cerebrale extracellulare e dell'L-aspartato. Poiché questi sono anche dipendenti dall'energia, la brusca perdita di energia dovuta all'interruzione del flusso sanguigno può alla fine rompere la membrana neuronale e astrogliale. Nei neuroni, la depolarizzazione della membrana contribuisce alla scarica vescicolare. Inoltre, la degradazione energetica può anche causare un cambiamento nella loro azione, quindi, provocando l'attivazione di L-glutammato e L-aspartato e di influenzare l'omeostasi ionica che può interrompere l'azione EAAT. L'attivazione di L-glutammato / L-aspartato contribuisce all'eccitotossicità attraverso l'eccessiva attivazione di iGluR di tipo NMDA come dimostrato dall'efficienza degli antagonisti NMDA in modelli animali di ischemia cerebrale transitoria.
Nel trauma cranico, il danno meccanico ai tessuti e la rottura della barriera emato-encefalica possono innescare la neurodegenerazione secondaria acuta, che, insieme alla neuroinfiammazione e allo stress ossidativo, è associata all'attivazione dell'L-glutammato dai compartimenti intracellulari e, quindi, all'eccitotossicità acuta. Inoltre, l'applicazione acuta dell'antagonista NMDA MK801 a seguito di trauma cranico migliora la perdita neuronale e le anomalie comportamentali a lungo termine, tra le altre.
Nello stato epilettico, la continuazione dell'attività sincronizzata delle reti neuronali eccitatorie così come la continua rottura dei meccanismi restrittivi è la principale fonte di attivazione di L-glutammato e L-aspartato. Poiché la gravità dell'attività sincrona dipende dal coinvolgimento delle cellule nervose in un sistema neuronale e la capacità di una cellula neurale di resistere al glutammato in eccesso dipende principalmente dal modello di espressione degli iGluR, una degenerazione delle popolazioni neuronali associata alla maturazione e alquanto limitata che è infine causato da crisi epilettiche prolungate. Il significato dell'eccitotossicità nello stato epilettico è dimostrato dal fatto che gli antagonisti NMDA, come la ketamina, riducono la perdita surrenalica.
Eccitotossicità nelle malattie neurologiche
Poiché è stato scoperto che gli EAAT sono sottoregolati in una varietà di problemi di salute del sistema nervoso centrale (SNC) e L-glutammato, così come L-aspartato, la clearance può in ultima analisi influenzare l'eccitotossicità delle malattie neurologiche, molti professionisti sanitari hanno deciso di determinare sostanze che causano EAAT2, o il principale EAAT nel cervello e più comunemente dimostrato di essere sottoregolato. Ciò ha dimostrato sostanze che mostrano l'espressione EAAT2 astrocitica sia in studi di ricerca in vitro che in vivo. Molti di questi hanno anche dimostrato proprietà protettive in modelli animali di malattie neurologiche. Cef è uno dei composti più valutati ed è stato analizzato in modelli di AD, HD e SLA con esiti positivi. Tuttavia, nessuna delle sostanze è stata ampiamente studiata per la sua capacità di interagire con altri percorsi neuroprotettivi. È stato anche dimostrato che Cef promuove l'espressione di EAAT2 ma anche di innescare il fattore di trascrizione Nrf2, che si traduce nella trascrizione di un'ampia gamma di geni coinvolti nella citoprotezione e nella protezione antiossidante. Poiché si ritiene che lo stress ossidativo svolga un ruolo essenziale in molte, se non tutte, malattie neurologiche, questo percorso può spiegare la neuroprotezione causata dalla Cef. Inoltre, xCT, che può essere uno dei bersagli a valle di Nrf2, ha dimostrato di essere sovraregolato da Cef in vitro e in vivo. Un'altra sostanza in vitro che promuove l'EAAT2, MS-153, protegge efficacemente dalla neurodegenerazione secondaria dopo una lesione cerebrale traumatica e attraverso meccanismi diversi dalla sovraregolazione EAAT2. La prova di esperimenti concettuali che dimostrano l'aumento della stimolazione tramite iGluR nelle malattie neurodegenerative necessita di manipolazioni della loro fisiologia dei neurotrasmettitori.
I topi Glud1 Tg dimostrano un modello di eccitotossicità associata a una maggiore attivazione sinaptica di L-glutammato con perdita neuronale limitata. Tuttavia, questo modello animale di neurotrasmissione glutamatergica non è stato ancora utilizzato per analizzare se la sovraespressione di Glud1 aggrava il fenotipo dei modelli murini nelle malattie neurologiche. Un'altra versione riguarda il mouse con deficit di EAAT2. I topi knock-out EAAT2 omozigoti hanno problemi di salute associati a morte prematura a causa di epilessia e atrofia corticale focale e ippocampale. I topi knock-out EAAT2 eterozigoti, tuttavia, si sviluppano normalmente e mostrano solo lievi anomalie comportamentali. Questo modello murino di iperfunzione moderata del glutammato è stato utilizzato in una raccolta di prove di studi di ricerca principali che hanno dimostrato il ruolo fondamentale del glutammato. I topi SLA, che hanno sia la mutazione mSOD93 G1A che una quantità ridotta di EAAT2 (SOD1 (G93A) / EAAT2 ), hanno rivelato un aumento della velocità di declino motorio accompagnato da una perdita precoce dei motoneuroni rispetto ai topi G93A mSOD1 Tg con mutante singolo . In questi topi mutanti è stata dimostrata anche una diminuzione della sopravvivenza. Quando incrociato con topi transgenici che esprimono mutazioni dell'amiloide umana-? precursore della proteina e presenilina-1 (A? PPswe / PS1? E9), perdita parziale dei deficit di memoria spaziale smascherati EAAT2 in topi di 6 mesi che esprimono A? PPswe / PS1? E9. Questi topi hanno dimostrato un aumento del rapporto di A? 42 / A? 40 insolubile in detergenti dimostrando che le carenze nella funzione del trasportatore del glutammato alla fine causano processi patogeni prematuri associati all'AD. In confronto, il fenotipo del modello di topo R6 / 2 HD non è stato modificato nei topi che avevano un solo allele EAAT2. Ulteriori studi di ricerca sono ancora necessari per ulteriori prove.
Come complemento a questi studi di ricerca, sono stati sviluppati anche topi transgenici che sovraesprimono EAAT2 negli astrociti attraverso il promotore GFAP. I topi a doppia Tg EAAT2 / G93A mSOD1 hanno dimostrato un moderato miglioramento del loro fenotipo simile alla SLA con un ritardo statisticamente significativo (14 volte) nella diminuzione della forza di presa e nella perdita dei motoneuroni, nonché una diminuzione in altre occasioni, come l'attivazione della caspasi-3 e SOD1, sebbene non all'inizio di paralisi, perdita di peso o allungamento della vita rispetto ai cuccioli monotransgenici G93A mSOD1. È stato utilizzato esattamente lo stesso modello di topo transgenico EAAT2 per valutare l'effetto di un migliore assorbimento astrocitario di L-glutammato e L-aspartato mediante incroci con un modello animale di topi AD, A? PPswe / Ind. L'aumento dei livelli di proteina EAAT2 ha notevolmente aumentato e migliorato il funzionamento cognitivo generale, ripristinato l'etica sinaptica e diminuito le placche amiloidi in quei topi AD.
Nei topi in cui la regolazione geneticamente modificata e la gestione di xCT causa una carenza nel sistema anti-glutammato / cistina x? C, l'ovvia diminuzione dell'L-glutammato extrasinaptico è associata all'enorme resistenza dei neuroni dopaminergici contro la neurodegenerazione indotta dalla 6-idrossidopamina, forse come conseguenza della ridotta eccitotossicità. Tuttavia, è stato anche dimostrato che l'attivazione della microglia è modulata da carenze del sistema x? C che portano a un fenotipo più neuroprotettivo che offre una spiegazione per l'effetto protettivo della delezione xCT in questa circostanza.
Pertanto, le variazioni genetiche incoraggiano il ruolo dell'eccitotossicità cronica nelle malattie neurodegenerative, in particolare AD e SLA. Tutti questi modelli rappresentano cambiamenti per tutta la vita nella neurotrasmissione glutamatergica. Questi modelli non possono determinare se l'utilizzo di farmaci e / o farmaci può influenzare direttamente i livelli di glutammato durante il processo neurodegenerativo e / o essere protettivo. Sia la valutazione che l'analisi della medicina inducente EAAT2 per la progressione di modelli murini inducibili e la loro interazione con altre vie di segnalazione sono ancora garantite da ricercatori e operatori sanitari.
In molti studi di ricerca, le prove e le misure di esito hanno dimostrato che la disregolazione del glutammato e l'eccitotossicità in molte malattie neurologiche, tra cui AD, HD e SLA, alla fine portano alla neurodegenerazione e a una varietà di sintomi associati ai problemi di salute. Lo scopo del seguente articolo è discutere e dimostrare il ruolo che la disregolazione del glutammato e l'eccitotossicità svolgono sulle malattie neurodegenerative. I meccanismi per l'eccitotossicità sono diversi per ogni problema di salute. - Dr. Alex Jimenez DC, CCST Insight - Dr. Alex Jimenez DC, CCST Insight
Modulo di valutazione metabolica
Il seguente modulo di valutazione metabolica può essere compilato e presentato al Dr. Alex Jimenez. I gruppi di sintomi elencati in questo modulo non devono essere utilizzati come diagnosi di alcun tipo di malattia, condizione o qualsiasi altro tipo di problema di salute.
L'eccitotossicità è caratterizzata come un insulto acuto che causa la morte cellulare a causa dell'eccessiva attivazione di iGluRs. L'eccitotossicità svolge un ruolo fondamentale in una varietà di problemi di salute del sistema nervoso centrale (SNC), tra cui ischemia cerebrale, TBI e stato epilettico. I meccanismi per l'eccitotossicità acuta sono diversi per ogni problema di salute. Lo scopo delle nostre informazioni è limitato a problemi di chiropratica, salute muscoloscheletrica e nervosa, nonché articoli, argomenti e discussioni di medicina funzionale. Utilizziamo protocolli sanitari funzionali per il trattamento di lesioni o disturbi cronici del sistema muscolo-scheletrico. Per discutere ulteriormente l'argomento di cui sopra, non esitate a chiedere al Dr. Alex Jimenez o contattarci al numero 915-850-0900 .
A cura del Dr. Alex Jimenez
Riferimenti ï ¿½
Lewerenz, Jan e Pamela Maher. Tossicità cronica del glutammato nelle malattie neurodegenerative: quali sono le prove? Frontiere in Neuroscienze, Frontiers Media SA, 16 dic. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
Discussione argomento aggiuntiva: dolore cronico
Il dolore improvviso è una risposta naturale del sistema nervoso che aiuta a dimostrare possibili lesioni. Ad esempio, i segnali del dolore viaggiano da una regione lesa attraverso i nervi e il midollo spinale al cervello. Il dolore è generalmente meno grave poiché la lesione guarisce, tuttavia, il dolore cronico è diverso dal tipo medio di dolore. Con dolore cronico, il corpo umano continuerà a inviare segnali di dolore al cervello, indipendentemente dal fatto che la lesione sia guarita. Il dolore cronico può durare da alcune settimane a persino diversi anni. Il dolore cronico può influenzare enormemente la mobilità di un paziente e può ridurre la flessibilità, la forza e la resistenza.
Zoomer Plus neuronale per la malattia neurologica
ï ¿½
Il dottor Alex Jimenez utilizza una serie di test per aiutare a valutare le malattie neurologiche. Lo zoom neuraleTM Plus è una gamma di autoanticorpi neurologici che offre un riconoscimento specifico anticorpo-antigene. Il vibrante Neural ZoomerTM Plus è progettato per valutare la reattività di un individuo a 48 antigeni neurologici con connessioni a una varietà di malattie neurologicamente correlate. Il vibrante zoomer neuraleTM Plus mira a ridurre le condizioni neurologiche fornendo ai pazienti e ai medici una risorsa vitale per la diagnosi precoce del rischio e una maggiore attenzione alla prevenzione primaria personalizzata.
Formule per il supporto alla metilazione
XYMOGEN s Le formule professionali esclusive sono disponibili tramite professionisti sanitari selezionati. La vendita e lo sconto su Internet delle formule XYMOGEN sono severamente vietati.
Orgogliosamente, Il dottor Alexander Jimenez rende le formule XYMOGEN disponibili solo per i pazienti sotto la nostra cura.
Si prega di chiamare il nostro ufficio in modo da poter assegnare una consulenza medica per l'accesso immediato.
Se sei un paziente di Injury Medical & Chiropractic Clinica, puoi chiedere informazioni su XYMOGEN chiamando 915-850-0900.
ï ¿½
Per vostra comodità e revisione del XYMOGEN prodotti si prega di rivedere il seguente link. *XYMOGEN-Catalog-Scaricare ï ¿½
* Tutte le precedenti politiche XYMOGEN rimangono rigorosamente in vigore.
Precedenti studi di ricerca suggeriscono che l'L-aspartato, come l'L-glutammato, innesca l'attività eccitatoria sui neuroni. L-aspartato funziona con L-glutammato nelle vescicole sinaptiche delle sinapsi eccitatorie asimmetriche. Ma la concentrazione totale di questi nel cervello umano (0.96-1.62? Mol / grammo di peso umido), le loro concentrazioni extracellulari nella corteccia misurate mediante microdialisi (1.62? M per L-aspartato e 9.06? M per L-glutammato) e il loro apporto secondo l'immunoistochimica suggeriscono che l'L-aspartato è significativamente meno abbondante dell'L-glutammato. Inoltre, l'L-aspartato è un potente agonista per i recettori NMDA ma non per altri iGluR con un EC50 solo otto volte superiore a quello dell'L-glutammato. Gli EAAT che svolgono un ruolo fondamentale nell'assorbimento di tutto l'L-glutammato rilasciato vescicolare nel sistema nervoso centrale (SNC) richiedono anche l'utilizzo dell'L-aspartato. L'L-aspartato è forse meno essenziale dell'L-glutammato connesso all'attività eccitatoria totale associata agli iGluRs. Insieme al suo ruolo di neurotrasmettitore, come accennato in precedenza, l'L-aspartato è anche necessario come substrato per l'aspartato amino-transferasi che si trasforma in 2-ossoglutarato e L-glutammato per il trasporto alle vescicole corticali dei neuroni glutamatergici che possono anche conseguentemente e aumentare indirettamente il rilascio di L-glutammato.
Altre molecole nella segnalazione di glutammato
Una caratteristica che distingue i recettori NMDA dai diversi iGluR è che l'attivazione dei recettori NMDA richiede la connessione di un co-agonista alla regione di legame della glicina del recettore. Ad esempio, nella retina e nel midollo spinale, l'origine della glicina può fuoriuscire dalle sinapsi inibitorie glicinergiche. Ma, in diverse regioni del cervello con una maggiore espressione del recettore NMDA, come la formazione dell'ippocampo, mancano le reazioni associate ai recettori della glicina sensibili alla stricnina, almeno nei neuroni adulti, a dimostrazione dell'assenza di neurotrasmissioni inibitorie glicinergiche. Ma la glicina si trova nel fluido extracellulare dell'ippocampo a quantità basali di circa 1.5 μM, che è simile alla saturazione della regione di legame della glicina del recettore NMDA, sebbene questi possano essere regolati verso l'alto e verso il basso. L'origine della glicina extracellulare nell'ippocampo può essere neuroni che rilasciano glicina attraverso il trasportatore di amminoacidi alanina-serina-cisteina 1 (asc-1). Ma è stato anche dimostrato il rilascio di glicina da parte degli astrociti stimolato dalla depolarizzazione e dal kainato. Sono necessari ulteriori studi di ricerca per mostrare alla fine queste misure di esito.
Anche in precedenti studi di ricerca sul recettore NMDA e sulla sua co-attivazione da parte della glicina è emerso che gli amminoacidi D, in particolare la D-serina, sono potenti quasi quanto la glicina. Solo diversi anni dopo, è diventato ovvio che la D-serina si trova nel cervello di ratti e umani a circa un terzo della loro concentrazione di L-serina con una concentrazione assoluta di oltre 0.2 μmol / g di tessuto cerebrale. Utilizzando un antisiero per la D-serina, studi di ricerca hanno dimostrato che la D-serina dal cervello si trova solo negli astrociti e la sua fornitura si adatta all'espressione dei recettori NMDA. Inoltre, gli stessi ricercatori hanno dimostrato che la D-serina viene rilasciata dagli astrociti coltivati quando esposti a L-glutammato o kainato. L'abbondanza di D-serina è trovata dall'enzima degradante D-amminoacido ossidasi (DAO) che rivela una maggiore espressione nel hindbrain dove i livelli di D-serina sono ridotti così come l'enzima sintetico serina racemasi che crea D-serina da L- serina. La D-serina sembra essere immagazzinata nelle vescicole citoplasmatiche degli astrociti e può essere rilasciata per esocitosi. Il potenziamento a lungo termine dipende dal rilascio di D-serina dagli astrociti nelle fettine dell'ippocampo, suggerendo che questo amminoacido gioca sicuramente un ruolo fondamentale nella neurotrasmissione glutamatergica attraverso i recettori NMDA. Inoltre, nelle fettine dell'ippocampo, studi di ricerca hanno trovato, utilizzando D-serina e enzimi degradanti della glicina, che D-serina funziona come co-trasmettitore per i recettori NMDA sinaptici sui neuroni CA1, allo stesso modo la glicina funziona come co-agonista endogeno per i recettori NMDA extrasinaptici. I recettori NMDA sinaptici dei neuroni del giro dentato utilizzano la glicina anziché la D-serina come co-agonista.
Prese collettivamente, le misure di esito multistrato mostrano che l'L-aspartato non funziona semplicemente come un agonista sui recettori NMDA, ma anche la glicina e la D-serina svolgono un ruolo fondamentale nella neurotrasmissione glutamatergica nel cervello umano. Ma anche altre molecole hanno dimostrato di essere importanti modulatori della neurotrasmissione glutamatergica.
Glutammato attivato da altre molecole
L-omocisteato (L-HCA) ha somiglianze strutturali con L-glutammato. L'amminoacido non proteico è un prodotto di ossidazione dell'omocisteina che viene biosintetizzato dalla metionina nell'eliminazione del proprio gruppo metilico terminale ed è anche un intermedio della via di transolfurazione mediante la quale la metionina può essere convertita in cisteina attraverso la cistationina. I primi studi di ricerca hanno dimostrato che questo amminoacido può causare l'afflusso di calcio nei neuroni coltivati in modo sicuro ed efficace come l'L-glutammato. Inoltre, L-HCA ha rivelato un'aumentata affinità per i recettori NMDA rispetto ad altri iGluR nei saggi di legame associati alla sua capacità di causare eccitotossicità inibibile dall'antagonista del recettore NMDA e afflusso di sodio. Inoltre, L-HCA può attivare mGluR5 con la stessa efficienza dell'L-glutammato. L-HCA si trova nel cervello, tuttavia, è stato dimostrato che le concentrazioni sono circa 500 volte inferiori a quelle dell'L-glutammato e persino 100 volte inferiori rispetto a quelle dell'L-aspartato in diverse regioni del cervello del ratto. Durante la stimolazione indotta dal potassio, la scarica di L-HCA viene attivata dalle preparazioni di fette di cervello come dimostrato per L-aspartato e L-glutammato sebbene il rilascio assoluto di HCA sia circa 50 volte inferiore. Sorprendentemente, l'HCA è un inibitore competitivo molto efficiente della cistina e dell'assorbimento di L-glutammato attraverso il sistema antiportatore di cistina / glutammato x? C, l'attività che regola e gestisce le concentrazioni extracellulari di L-glutammato extrasinaptico nel cervello. Pertanto, l'impatto di L-HCA sull'attivazione di NMDA e di altri recettori di L-glutammato può anche fare affidamento sull'innesco di L-glutammato indotto da L-HCA attraverso il sistema x? C. L-HCA può svolgere un ruolo importante nella stimolazione complessiva dei recettori L-glutammato. Tuttavia, questo può cambiare enormemente in determinate condizioni, p. Es., Nei pazienti con terapia ad alte dosi con metotrexato, un farmaco antitumorale che, limitando la diidrofolato reduttasi, limita il riciclo catalizzato dal tetraidrofolato della metionina dall'omocisteina. Qui, concentrazioni di L-HCA superiori a 100 μM sono state dimostrate dal liquido cerebrospinale mentre L-HCA non era rilevabile nei soggetti di controllo. Sono ancora necessari ulteriori studi di ricerca per determinare queste misure di esito.
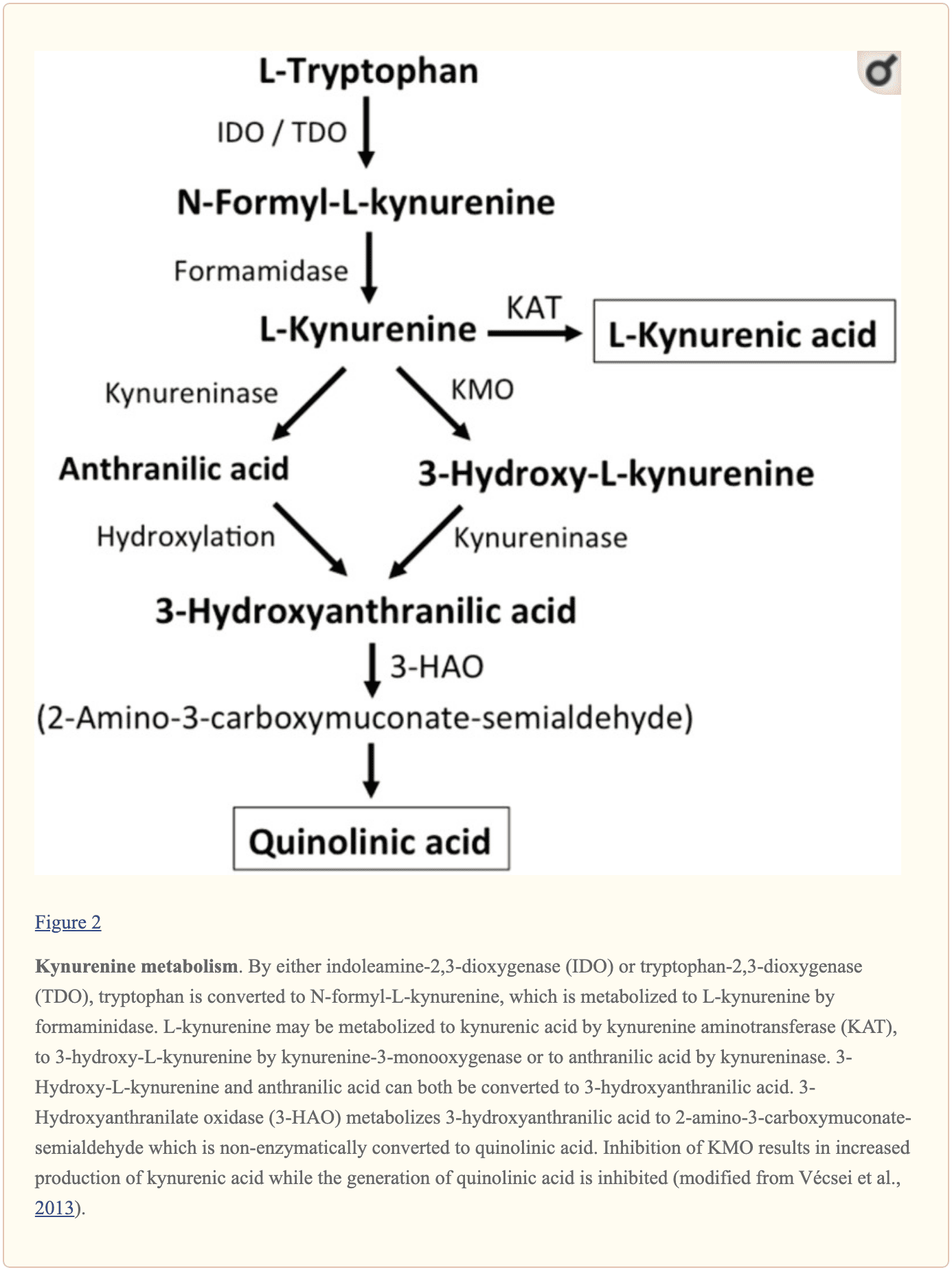
Ulteriori piccole molecole endogene che si ritiene influenzino la segnalazione di L-glutammato includono diversi intermedi del metabolismo del triptofano, come mostrato nella Figura 2. Attraverso l'attività dell'indoleamina 2,3-diossigenasi (IDO) o triptofano 2,3-diossigenasi (TDO), il triptofano viene trasformato in N-formil-L-chinurenina che viene successivamente trasformata in chinurenina (KYN) dalla formamidasi. Tre percorsi, due dei quali si collegano in una fase successiva, determinano un ulteriore metabolismo. Innanzitutto, attraverso l'attività della chinurenina aminotransferasi (KAT), il KYN viene convertito in acido chinurenico (KYNA). KYN può anche essere convertito in 3-idrossichinurenina (3HK) dalla chinurenina monoossigenasi (KMO), che può essere successivamente utilizzata come substrato dalla chinureninasi per la sintesi dell'acido 3-idrossiantranilico (3HANA). Inoltre, utilizzando KYN come substrato, la chinureninasi sviluppa acido antranilico (ANA), che mediante idrossilazione aspecifica può anche essere convertito in 3HANA. Secondo studi di ricerca, 3HANA funziona finalmente come substrato per la generazione di acido chinolinico (QUIN).
La concentrazione di triptofano nel cervello del ratto è di circa 25 nmol / g di peso umido e circa 400 volte inferiore all'L-glutammato e 100 volte inferiore all'L-aspartato. I livelli cerebrali dimostrati di chinurenine sono ancora più bassi con 0.4-1.6 nmol / g per QUIN, 0.01-0.07 nmol / ml per KYNA e 0.016 nmol / g per 3HANA. Circa il 40% del KYN cerebrale è sintetizzato localmente. I metaboliti del triptofano dimostrano un legame differenziale con le proteine plasmatiche e il loro trasporto attraverso la barriera che è molto diverso. KYN e 3HK vengono trasportati attraverso il grande sistema di trasporto di amminoacidi neutri L. Le kynurenine sembrano penetrare nel cervello umano per diffusione passiva. Inoltre, KYNA, 3HANA e soprattutto ANA si legano alle proteine del siero che alla fine ne restringono e limitano la diffusibilità attraverso la barriera ematoencefalica.
Studi di ricerca hanno dimostrato che QUIN, quando utilizzato ionoforeticamente nelle cellule di ratto, ha causato l'attivazione neuronale che è stata prevenuta da un antagonista del recettore NMDA, suggerendo che QUIN può funzionare come un agonista del recettore NMDA. Tuttavia, è stato dimostrato che l'EC50 per QUIN per attivare le correnti del recettore NMDA è circa 1000 volte superiore all'EC50 dell'L-glutammato. È stato dimostrato che l'iniezione intracerebrale di QUIN causa cambiamenti ultrastrutturali, neurochimici e comportamentali simili a quelli causati dagli agonisti del recettore NMDA. Il fatto che le concentrazioni di QUIN siano da circa 5000 a 15,000 volte inferiori alle concentrazioni di L-glutammato cerebrale rende improbabile che la modulazione della segnalazione del recettore NMDA da parte di QUIN svolga un ruolo essenziale. È stato dimostrato che KYNA funziona come un antagonista del recettore NMDA. Ma, sebbene l'infusione con l'inibitore di KMO Ro 61-8048 abbia migliorato di 10 volte le concentrazioni di KYNA extracellulare cerebrale, ciò non ha comportato un'inibizione della depolarizzazione neuronale mediata da NMDA, una scoperta che sfida la convinzione che KYNA a quantità quasi fisiologiche direttamente modula i recettori NMDA. In confronto, l'aumento di KYNA nel cervello indotto dall'inibitore KMO JM6 ha diminuito la concentrazione di L-glutammato cerebrale extracellulare. Inoltre, i livelli di KYNA dal liquido cerebrale extracellulare sono stati associati ai livelli di L-glutammato suggerendo che anche a livelli fisiologici o vicini a quelli fisiologici, KYNA modula il metabolismo dell'L-glutammato. Sia l'attivazione del recettore accoppiato alla proteina G GPR35 che l'inibizione dei recettori nicotinici dell'acetilcolina presinaptica? 7 sono suggerite nella riduzione indotta da KYNA del rilascio di L-glutammato. Per riassumere, sebbene QUIN e L-HCA siano presenti nel cervello umano, le loro concentrazioni discutono contro di loro con ruoli nella regolazione e nel mantenimento della neurotrasmissione. Al contrario, anche se i percorsi devono essere definiti in maggiore dettaglio, le prove supportano i livelli e l'opinione che la scarica possa essere modulata da KYNA e neurotrasmissione.
Il glutammato, insieme all'aspartato e ad altre molecole, sono alcuni dei principali neurotrasmettitori eccitatori nel cervello umano. Sebbene questi svolgano un ruolo fondamentale nella struttura generale e nella funzione del sistema nervoso centrale, compresi il cervello e il midollo spinale, quantità eccessive di altre molecole possono in definitiva innescare i recettori del glutammato. L'eccesso di glutammato può causare eccitotossicità che può portare a una varietà di problemi di salute, come il morbo di Alzheimer e altri tipi di malattie neurologiche. Il seguente articolo descrive come altre molecole possono attivare i recettori del glutammato. - Dr. Alex Jimenez DC, CCST Insight - Dr. Alex Jimenez DC, CCST Insight
Studi di ricerca suggeriscono che L-aspartato, come il L-glutammato, innesca l'attività eccitatoria. L-aspartato funziona con L-glutammato nelle vescicole sinaptiche delle sinapsi eccitatorie asimmetriche. Ma la concentrazione totale di questi nel cervello umano suggerisce che L-aspartato è significativamente meno abbondante di L-glutammato. Inoltre, L-aspartato è un potente agonista per i recettori NMDA ma non per altri iGluR con un EC50 appena otto volte superiore a quello del L-glutammato. Lo scopo delle nostre informazioni è limitato a problemi di chiropratica, salute muscoloscheletrica e nervosa, nonché articoli, argomenti e discussioni di medicina funzionale. Utilizziamo protocolli sanitari funzionali per il trattamento di lesioni o disturbi cronici del sistema muscolo-scheletrico. Per discutere ulteriormente l'argomento di cui sopra, non esitate a chiedere al Dr. Alex Jimenez o contattarci al numero 915-850-0900 .
A cura del Dr. Alex Jimenez
Riferimenti ï ¿½
Lewerenz, Jan e Pamela Maher. Tossicità cronica del glutammato nelle malattie neurodegenerative: quali sono le prove? Frontiere in Neuroscienze, Frontiers Media SA, 16 dic. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
Discussione argomento aggiuntiva: dolore cronico
Il dolore improvviso è una risposta naturale del sistema nervoso che aiuta a dimostrare possibili lesioni. Ad esempio, i segnali del dolore viaggiano da una regione lesa attraverso i nervi e il midollo spinale al cervello. Il dolore è generalmente meno grave poiché la lesione guarisce, tuttavia, il dolore cronico è diverso dal tipo medio di dolore. Con dolore cronico, il corpo umano continuerà a inviare segnali di dolore al cervello, indipendentemente dal fatto che la lesione sia guarita. Il dolore cronico può durare da alcune settimane a persino diversi anni. Il dolore cronico può influenzare enormemente la mobilità di un paziente e può ridurre la flessibilità, la forza e la resistenza.
Zoomer Plus neuronale per la malattia neurologica
ï ¿½
Il dottor Alex Jimenez utilizza una serie di test per aiutare a valutare le malattie neurologiche. Lo zoom neuraleTM Plus è una gamma di autoanticorpi neurologici che offre un riconoscimento specifico anticorpo-antigene. Il vibrante Neural ZoomerTM Plus è progettato per valutare la reattività di un individuo a 48 antigeni neurologici con connessioni a una varietà di malattie neurologicamente correlate. Il vibrante zoomer neuraleTM Plus mira a ridurre le condizioni neurologiche fornendo ai pazienti e ai medici una risorsa vitale per la diagnosi precoce del rischio e una maggiore attenzione alla prevenzione primaria personalizzata.
Formule per il supporto alla metilazione
XYMOGEN s Formule professionali esclusive sono disponibili attraverso selezionati professionisti sanitari autorizzati. La vendita su Internet e l'attualizzazione delle formule XYMOGEN sono severamente vietate
Orgogliosamente, Il dottor Alexander Jimenez rende le formule XYMOGEN disponibili solo per i pazienti sotto la nostra cura.
Si prega di chiamare il nostro ufficio in modo da poter assegnare una consulenza medica per l'accesso immediato.
Se sei un paziente di Injury Medical & Chiropractic Clinica, puoi chiedere informazioni su XYMOGEN chiamando 915-850-0900.
ï ¿½
Per vostra comodità e revisione del XYMOGEN prodotti si prega di rivedere il seguente link. *XYMOGEN-Catalog-Scaricare ï ¿½
* Tutte le precedenti politiche XYMOGEN rimangono rigorosamente in vigore.
L'eccitotossicità è un meccanismo patologico visto in una varietà di problemi di salute in cui un'eccitazione sinaptica eccessiva causa la morte neuronale e si ritiene anche che sia causata dall'accumulo extracellulare del neurotrasmettitore eccitatorio glutammato, che innesca e collega l'N-metil-D-aspartato glutammatergico ionotropico recettori (NMDAR) nel cervello. In generale, gli NMDAR regolano e mantengono il calcio nelle cellule per aiutare a gestire i meccanismi fisiologici come la plasticità sinaptica e la memoria, tuttavia, una stimolazione eccessiva può alla fine aumentare il calcio intracellulare che innesca la segnalazione di morte cellulare per attivare l'apoptosi. Questo meccanismo patologico è stato suggerito in una varietà di problemi di salute, come lesioni cerebrali traumatiche (TBI) e malattia di Alzheimer (AD), dove viene ampiamente esaminato per comprendere i problemi di salute e gli approcci terapeutici. In un ictus, l'eccitotossicità ha dimostrato di essere il principale meccanismo patologico in cui si verifica il danno neuronale ed è considerato un obiettivo ben noto per molti recenti tentativi di sviluppo di terapie per l'ictus.
L'ictus è un problema acuto di salute del cervello che causa danni neuronali che attualmente non ha approcci di trattamento neuroprotettivo sicuro ed efficace. Immediatamente dopo un ictus, il tessuto cerebrale perde la perfusione sanguigna e il centro dell'infarto si deteriora rapidamente. Ciò provoca quindi un'ischemia più lieve e molte cellule cerebrali o neuroni si tradurrà in una morte ritardata che può richiedere fino a diverse ore o addirittura giorni. Studi di ricerca mostrano che il meccanismo della morte cellulare è principalmente l'eccitotossicità dipendente dal recettore NMDA. Nelle aree ischemiche, i livelli di glutammato extracellulare aumentano prevenendo il rilascio di glutammato, l'attività sinaptica o l'attivazione NMDAR che era in grado di limitare la morte cellulare in una varietà di modelli di ictus. Pertanto, prevenire l'eccitotossicità è un approccio terapeutico importante per ridurre il danno cerebrale e migliorare le misure di esito del paziente a seguito di un ictus, e questo ha sicuramente incoraggiato ampi sforzi verso lo sviluppo di approcci di trattamento dell'ictus basati sul recettore NMDA negli ultimi due decenni. Purtroppo, questi hanno in gran parte ottenuto risultati piuttosto deludenti. Diversi studi di ricerca non sono riusciti a trovare l'efficienza attesa di NMDAR per ridurre le lesioni cerebrali. Le ragioni alla base dei risultati degli studi di ricerca di base e degli studi clinici sono ancora sconosciute, tuttavia sono state suggerite diverse ragioni. Questi includono, ma non sono limitati a, l'incapacità di utilizzare le dosi corrette necessarie per la neuroprotezione a causa dei loro effetti collaterali, l'incapacità di usare i farmaci entro le loro finestre neuroprotettive, i disegni sperimentali poveri e l'eterogeneità nella popolazione dei pazienti. Tuttavia, come riassumeremo brevemente nel seguente articolo, il miglioramento della nostra comprensione dei meccanismi fisiologici e patologici dell'attivazione NMDAR nonché dei diversi percorsi collegati a diversi sottotipi NMDAR, ha permesso ai ricercatori di sviluppare nuovi approcci terapeutici che migliorano le finestre terapeutiche e aumentare la specificità per le vie di segnalazione della morte, ottenendo la neuroprotezione senza interrompere altre vie di segnalazione essenziali a valle del recettore NMDAR.
Neuroprotectants Targeting Sottotipi NMDAR
I sottotipi NMDAR hanno scopi diversi in termini di eccitotossicità e fisiologia. Il NMDAR è un recettore che generalmente ha due subunità GluN1, note anche come NR1, nonché due subunità della sottofamiglia GluN2 (GluN2A-2D, noto anche come NR2A-2D). Nella corteccia, le principali sottopopolazioni di NMDAR sono i recettori contenenti GluN2A o GluN2A e 2B. I recettori contenenti GluN2A si trovano nelle sinapsi mentre i recettori contenenti GluN2B si trovano sulle membrane extrasinaptiche. I recettori contenenti GluN2A e GluN2B sono diversi l'uno dall'altro perché regolano e gestiscono la plasticità, favorendo il potenziamento a lungo termine (GluN2A) o la depressione (GluN2B) attraverso una varietà di proprietà elettrofisiologiche e farmacologiche nonché proteine di segnalazione. Inoltre, questi recettori svolgono un ruolo fondamentale nel promuovere la sopravvivenza cellulare (GluN2A) o la morte (GluN2B) dopo stimolazione eccitotossica. Poiché i recettori contenenti GluN2A sono principalmente focalizzati sulle sinapsi mentre i recettori contenenti GluN2B sono focalizzati sulle membrane sinaptiche ed extrasinaptiche, quando le condizioni eccitotossiche fanno sì che il glutammato si estenda oltre le sinapsi, la segnalazione di morte mediata da GluN2B diventa più forte rispetto alla segnalazione di sopravvivenza che alla fine si traduce in Morte. Attraverso un ictus, ad esempio, gli NMDAR hanno meno probabilità di favorire la sopravvivenza cellulare e possono invece causare effetti dannosi prevenendo considerevoli normali scopi fisiologici. Selfotel, un bloccante NMDAR non specifico, era neuroprotettivo contro l'ictus in vitro e in vivo, tuttavia, alla fine, non è riuscito a essere neuroprotettivo contro l'ictus negli studi clinici causando una serie di effetti collaterali intollerabili.
Le strategie di trattamento per ridurre gli effetti collaterali indesiderati, inclusi gli antagonisti del sito della glicina e miglioramenti specifici del sottotipo NMDAR, erano di indirizzare le regioni di legame della glicina allosterica sulle subunità GluN1 con licostinel e gavestinel invece di bloccare direttamente il recettore. Questi farmaci candidati hanno ottenuto buoni risultati negli esami preclinici, tuttavia, hanno anche fallito a causa della bassa efficienza nonostante i profili di effetti collaterali minimi. Gli effetti collaterali negativi erano forse dovuti a una finestra di tempo mancata a seguito di un ictus che mostra quali bloccanti del recettore sono sicuri ed efficaci nel prevenire la morte.
Migliori metodi e tecniche di trattamento per ridurre gli effetti collaterali indesiderati di NMDAR devono utilizzare le differenze tra le loro variazioni. Ad esempio, l'inibitore specifico di GluN2B traxoprodil è neuroprotettivo negli studi di ricerca sull'ictus e ha effetti collaterali minimi, tuttavia, ha anche fallito negli studi clinici. Simile agli antagonisti della regione della glicina, forse deve essere adeguatamente regolato e gestito per funzionare in modo efficiente. Gli agonisti di GluN2A dovrebbero promuovere la segnalazione di sopravvivenza cellulare che potrebbe consentire il recupero dopo un ictus e la sopravvivenza cellulare per impedire il passaggio di segnalazione. In effetti, l'attivazione dei recettori contenenti GluN2A utilizzando dosi aumentate di glicina era neuroprotettiva in un modello animale di ictus, ma ulteriori studi di ricerca devono esaminare l'attivazione di GluN2A come approccio terapeutico nei partecipanti umani.
Mentre gli antagonisti e i modulatori NMDAR sono sicuri ed efficaci nell'attenuare l'eccitotossicità nelle versioni sperimentali, il loro difetto è la sfida nell'implementare approcci terapeutici precocemente in coincidenza con il culmine del rilascio di glutammato eccitotossico. I pazienti colpiti da ictus spesso non hanno alcuna possibilità di ricevere questi approcci terapeutici in tempo. Tuttavia, il problema di salute può essere evitato se i bloccanti del recettore possono essere utilizzati nelle popolazioni a rischio. Uno studio di ricerca ha dimostrato che basse dosi di memantina profilattica, un antagonista NMDAR non competitivo con pochi effetti collaterali, possono ridurre considerevolmente le lesioni cerebrali e i deficit funzionali a seguito di un ictus. Resta da dimostrare se i farmaci siano tollerabili, sicuri ed efficaci se assunti in questo modo, ma soluzioni innovative potrebbero comunque affrontare il modo di somministrare quei farmaci.
Un fattore oltre a quelli degli studi clinici falliti è l'interazione degli NMDAR nella sopravvivenza cellulare che può essere completamente fraintesa. Negli ultimi decenni, sono state accumulate prove che gli NMDAR sinaptici possono anche causare la morte cellulare e GluN2A, così come GluN2B, non hanno necessariamente funzioni dicotomiche nell'eccitotossicità. Potrebbero essere necessari ulteriori studi di ricerca per dimostrare strategie più sfumate per gli inibitori dei recettori e per risolvere questa controversia.
Neuroprotettori che prendono di mira la segnalazione di morte cellulare
Un approccio terapeutico per gli inibitori NMDAR consiste nel concentrarsi sugli eventi più a valle della morte cellulare che si verificano in un periodo di tempo molto più lungo dopo l'attivazione del recettore. È stata determinata una varietà di percorsi di morte cellulare a seguito dell'attivazione e diversi gruppi hanno fornito prove di principio che questi percorsi possono essere regolati e gestiti con l'utilizzo di peptidi per proteggere in ultima analisi le cellule cerebrali oi neuroni senza effetti collaterali.
La più antica strategia peptidica segnalata e più esplorata negli obiettivi dell'ictus è la morte cellulare mediata dal protossido di azoto (nNOS). NNOS si collega alla proteina postsinaptica 95 (PSD95) che poi si collega alla coda C-terminale della subunità GluN2B. NOS è un enzima attivato dal calcio che attiva lo sviluppo dell'ossido nitrico (NO) e il proprio stato nel complesso recettore che lo associa in prossimità del flusso focalizzato di calcio che entra in GluN2B attivato. In un ictus, l'eccessivo afflusso di calcio attiva nNOS accoppiati a GluN2B. Un peptide di interferenza viene utilizzato per scollegare il complesso e prevenire lo sviluppo di NO. Il peptide, Tat-NR2B9c, è costituito da una sequenza di penetrazione cellulare derivata da Tat dell'HIV-1 che consente il passaggio attraverso la barriera ematoencefalica e le membrane cellulari, collegata a una copia della regione sul GluN2B per PSD95. Il peptide e GluN2B disconnettono PSD95, quindi, disaccoppiando nNOS nei livelli considerevoli di calcio locali senza interrompere la funzione del recettore da diverse vie. L'utilizzo si traduce in una notevole protezione contro i danni tissutali e funzionali senza effetti collaterali in vitro e in vivo dopo una singola dose somministrata prima o dopo l'ischemia in vivo. Il peptide è recentemente riuscito nella sperimentazione clinica di fase II in cui ha ridotto gli infarti iatrogeni durante il trattamento dell'aneurisma intracranico. Questa è la prima volta che uno studio di ricerca ha dimostrato l'efficienza negli esseri umani che mostra anche l'autenticità che il targeting della morte cellulare a valle può essere utile contro le lesioni neuronali eccitotossiche / ischemiche.
Mentre l'utilizzo dei peptidi in un ambiente clinico è sicuro ed efficace, un'efficienza simile è stata ottenuta con farmaci a piccole molecole che agiscono esattamente per lo stesso obiettivo e funzionano come i peptidi in un ambiente di laboratorio. Per imitare Tat-NR2B9c, è stato dimostrato individualmente che due piccole molecole, IC87201 e ZL006, competono nella regione di connessione specifica di GluN2B identica senza influenzare la connessione di PSD95 ad altre proteine. Inoltre, ZL006 imita la neuroprotezione del peptide senza causare notevoli effetti collaterali negativi. Identificando gli obiettivi e le regioni specifiche, gli studi di ricerca possono simulare farmaci a piccole molecole e accelerare la loro scoperta verso l'eccitotossicità e l'ictus.
Altri percorsi specifici per GluN2B sono stati dimostrati in modo simile e si stanno dimostrando promettenti nelle fasi di sviluppo. Uno di questi percorsi che viene attivato dopo l'attivazione di GluN2B è il potenziamento e il reclutamento di GluN2B nella membrana cellulare da parte della protein chinasi 1 associata alla morte (DAPK1). DAPK1 è una proteina che si collega alla calmodulina per attivare l'apoptosi ma è fosforilata in una forma inattiva che è incapace di associare morte cellulare e calmodulina. A seguito dell'eccitotossicità, l'attivazione della calcineurina defosforila e innesca DAPK1, contribuendo alla morte cellulare. Inoltre, il DAPK1 attivo può connettersi e fosforilare la coda C-terminale dei recettori, l'eccitotossicità e la loro funzione, aggravando l'afflusso di calcio. Un peptide di interferenza collegato a Tat che ha la regione di fosforilazione della coda C che è GluN2B è riuscito a bloccare l'interazione di DAPK1 attivo con GluN2B e promuovere l'eccitotossicità. Una volta che il peptide è stato utilizzato nei topi, soprannominato Tat-NR2B-CT, ha migliorato il risultato dopo l'ischemia. Tuttavia, Tat-NR2B-CT era efficace solo nel prevenire l'attività e l'inserimento incontrollato invece dell'apoptosi a valle della segnalazione DAPK1. I ricercatori sono stati anche in grado di collegare e guidare DAPK1 verso i lisosomi includendo una sequenza nella chiusura del peptide di ostacolo per creare un peptide di degradazione. Il risultato è stato un calo grave e temporaneo dei livelli di DAPK1 occupati con una corrispondente diminuzione dell'infarto durante la somministrazione del peptide ore dopo l'ischemia, secondo diversi studi di ricerca.
La chinasi 3 c-Jun N-terminale (JNK) agisce su molte vie ed è un mediatore per la morte cellulare nell'eccitotossicità. La proteina interagente JNK (JIP) collega e impedisce l'attività JNK attraverso un dominio di legame JNK (JBD) che si estende su oltre 20 residui. Quando questi residui sono collegati a Tat come dal peptide interrotto Tat-JBD20, sono in grado di limitare l'attività JNK e prevenire la morte cellulare nei modelli di ictus quando somministrati prima o dopo l'ischemia. È stato anche dimostrato che il peptide Tat-JBD20 utilizza D-amminoacidi invece di L-amminoacidi per resistere alla degradazione da parte delle proteasi endogene. Ciò aumenta enormemente l'emivita del peptide e non influisce negativamente sulla sua affinità di legame e selettività, dimostrando che questa alterazione può essere utilizzata per diversi peptidi di interferenza per aumentare l'efficienza e la biodisponibilità.
Vengono sempre scoperti nuovi obiettivi. Sebbene attualmente non vengano utilizzati nuovi approcci per il trattamento dell'ictus, sono stati compiuti molti progressi prendendo di mira i processi che si verificano durante l'ictus verso la creazione di approcci terapeutici. Con il debutto del raggiungimento di peptidi di degradazione e interruzione che prendono di mira eventi di segnalazione di passaggio specifici per GluN2B, c'è speranza che nuovi trattamenti siano all'orizzonte per problemi di salute che hanno eccitotossicità.
L'eccitotossicità è il meccanismo patologico mediante il quale le cellule cerebrali o i neuroni vengono infine danneggiati o eliminati dall'eccessiva stimolazione dei neurotrasmettitori, tra cui il glutammato e altre sostanze simili. Ciò si verifica in definitiva quando il recettore NMDA e il recettore AMPA sono iperattivati dai recettori del glutammato neurotrasmettitore eccitatorio. Ciò può causare una varietà di processi che possono danneggiare le strutture cellulari, compresi i componenti del citoscheletro, della membrana e del DNA. Regolamentare e gestire l'eccitotossicità può aiutare a mantenere il benessere generale. - Dr. Alex Jimenez DC, CCST Insight
L'eccitotossicità è un meccanismo patologico in cui un'eccitazione sinaptica eccessiva provoca la morte neuronale e si ritiene anche sia causata dall'accumulo extracellulare del neurotrasmettitore eccitatorio glutammato, che innesca e collega i recettori glutamatergici ionotropici N-metil-D-aspartato nel cervello (NMDAR) . Questo meccanismo patologico è stato suggerito in una varietà di problemi di salute, come la lesione cerebrale traumatica (TBI) e la malattia di Alzheimer (AD), dove è ampiamente esaminato per comprendere i problemi di salute e gli approcci terapeutici. Lo scopo delle nostre informazioni è limitato a problemi di chiropratica, salute muscoloscheletrica e nervosa, nonché articoli, argomenti e discussioni di medicina funzionale. Per discutere ulteriormente l'argomento di cui sopra, non esitate a chiedere al Dr. Alex Jimenez o contattarci al numero 915-850-0900 .
A cura del Dr. Alex Jimenez
Riferimenti ï ¿½
Li, Victor e Yu Tian Wang. Meccanismi molecolari dell'eccitotossicità mediata dal recettore NMDA: implicazioni per le terapie neuroprotettive per l'ictus. Ricerca sulla rigenerazione neuronale, Medknow Publications & Media Pvt Ltd, novembre 2016, www.ncbi.nlm.nih.gov/pmc/articles/PMC5204222/.
Discussione argomento aggiuntiva: dolore cronico
Il dolore improvviso è una risposta naturale del sistema nervoso che aiuta a dimostrare possibili lesioni. Ad esempio, i segnali del dolore viaggiano da una regione lesa attraverso i nervi e il midollo spinale al cervello. Il dolore è generalmente meno grave poiché la lesione guarisce, tuttavia, il dolore cronico è diverso dal tipo medio di dolore. Con dolore cronico, il corpo umano continuerà a inviare segnali di dolore al cervello, indipendentemente dal fatto che la lesione sia guarita. Il dolore cronico può durare da alcune settimane a persino diversi anni. Il dolore cronico può influenzare enormemente la mobilità di un paziente e può ridurre la flessibilità, la forza e la resistenza.
Zoomer Plus neuronale per la malattia neurologica
ï ¿½
Il dottor Alex Jimenez utilizza una serie di test per aiutare a valutare le malattie neurologiche. Lo zoom neuraleTM Plus è una gamma di autoanticorpi neurologici che offre un riconoscimento specifico anticorpo-antigene. Il vibrante Neural ZoomerTM Plus è progettato per valutare la reattività di un individuo a 48 antigeni neurologici con connessioni a una varietà di malattie neurologicamente correlate. Il vibrante zoomer neuraleTM Plus mira a ridurre le condizioni neurologiche fornendo ai pazienti e ai medici una risorsa vitale per la diagnosi precoce del rischio e una maggiore attenzione alla prevenzione primaria personalizzata.
Formule per il supporto alla metilazione
XYMOGEN s Le formule professionali esclusive sono disponibili tramite professionisti sanitari selezionati. La vendita e lo sconto su Internet delle formule XYMOGEN sono severamente vietati.
Orgogliosamente, Il dottor Alexander Jimenez rende le formule XYMOGEN disponibili solo per i pazienti sotto la nostra cura.
Si prega di chiamare il nostro ufficio in modo da poter assegnare una consulenza medica per l'accesso immediato.
Se sei un paziente di Injury Medical & Chiropractic Clinica, puoi chiedere informazioni su XYMOGEN chiamando 915-850-0900.
ï ¿½
Per vostra comodità e revisione del XYMOGEN prodotti si prega di rivedere il seguente link. *XYMOGEN-Catalog-Scaricare ï ¿½
* Tutte le precedenti politiche XYMOGEN rimangono rigorosamente in vigore.
Lo strumento Find A Practitioner di IFM è la più grande rete di riferimento in Medicina Funzionale, creata per aiutare i pazienti a localizzare professionisti di Medicina Funzionale in qualsiasi parte del mondo. I professionisti certificati IFM sono elencati per primi nei risultati di ricerca, data la loro vasta formazione in Medicina Funzionale
Prenotazione online e appuntamenti 24 ore su 7, XNUMX giorni su XNUMX *