Rispetto ad altri problemi di salute del sistema nervoso centrale (SNC), le malattie neurodegenerative croniche possono essere molto più complicate. In primo luogo, poiché la funzione mitocondriale compromessa è stata dimostrata in molte malattie neurodegenerative, i problemi risultanti nelle fonti di energia non sono così gravi come il collasso energetico nell'ictus ischemico. Pertanto, se l'eccitotossicità contribuisce alla neurodegenerazione, è necessario ipotizzare un tempo diverso di eccitotossicità cronica. Nel seguente articolo, illustreremo ciò che è noto sui percorsi che possono causare eccitotossicità nelle malattie neurodegenerative. Discuteremo specificamente che nella sclerosi laterale amiotrofica (SLA), nella malattia di Alzheimer (AD) e nella malattia di Huntington (HD) come esempi fondamentali con modelli animali sufficientemente convalidati in studi di ricerca.
Malattia di Huntington
La malattia di Huntington (MH) è una malattia neurodegenerativa fatale ereditaria causata da un'espansione ripetuta di trinucleotidi (CAG) nella regione codificante del gene huntingtin (Htt) che è associata alla degenerazione dei neuroni spinosi GABAergici di medie dimensioni ( MSN) nello striato, sebbene alla fine anche altre regioni del cervello possano essere colpite con il progredire del problema di salute. La MH è identificata come un disturbo del movimento con sintomatologia cognitiva e psichiatrica co-morbosa. Si ritiene che sia l'RNA mutante dell'htt sia la proteina codificata che include un'espansione ripetuta di poliglutammina causino i complicati cambiamenti nel metabolismo cellulare che si verificano nella disfunzione mitocondriale e nello stress ossidativo.
I primi risultati di studi di ricerca che hanno dimostrato che l'eccitotossicità può svolgere un ruolo fondamentale nella MH si basavano sull'osservazione che un'iniezione del loro metabolita KYN e dell'agonista del recettore NMDA QUIN, oltre a L-glutammato e kainato, nello striato dei ratti causava la degenerazione neuronale. . Un altro studio di ricerca ha stabilito che QUIN, rispetto a NMDA e kainato, causa la degenerazione selettiva degli MSN invece della morte neuronale, che assomiglia enormemente alla patologia della MH. Inoltre, i recettori NMDA hanno dimostrato di essere neuroni iperattivi e striatali da diversi modelli di topo HD, come un cromosoma artificiale di lievito (YAC) che porta alla sovraespressione di Htt a tutta lunghezza con ripetizioni di poliglutamine allungate e topi R6 / 2 è stato dimostrato che l'esone 1 di Http sovraesprimente con ripetizioni di poliglutammina allungate oltre a topi knock-in con ripetizioni CAG maggiori inserite dal gene Htt di topo, è stato sensibilizzato all'eccitotossicità in vitro. Inoltre, in vivo, una sensibilizzazione a un'iniezione di eccitotossina nello striato è stata dimostrata solo nel modello YAC transgenico di HD, mentre i topi con sovraesprimono i topi mutanti di esone 1, R6 / 1 e R6 / 2, o topi N171-82Q che sovraesprimono l'esone mutante 1 e i componenti dell'esone 2 o il cosiddetto topo "corto" che esprime la Htt N-terminale umana codificata dall'esone 1 e 2 con una ripetizione di 128 CAG al di sotto del promotore di Htt, ha prodotto una certa resistenza all'iniezione di eccitotossina striatale durante il processo di invecchiamento . Questa neuroprotezione non è necessariamente per gli agonisti del recettore NMDA, tuttavia, può aiutare diversi insulti neurotossici e può essere una risposta adattativa allo stress cellulare.
Il rilascio di MSN nel ratto ha aumentato i livelli di recettori NMDA contenenti NR2A e NR2B rispetto agli interneuroni nello striato. È stato dimostrato che l'espressione dell'mRNA di NR1 e NR2B nel neostriato dei pazienti con MH diminuisce notevolmente, il che è associato alla perdita di questi neuroni. Inoltre, è stato determinato che le vie mediate dal recettore NMDA in MSN sono estremamente sensibili all'inibitore specifico di NR2B ifenprodil. Nelle cellule HEK293, la sovraespressione del mutante HTTP ha aumentato le vie mediate dal recettore NMDA e ha aggravato il rilascio delle cellule indotto da NMDA solo quando NR2B- ma non quando i recettori NMDA contenenti NR2A erano co-espressi. Una possibile spiegazione per l'aumento dell'espressione del recettore NMDA contenente NR2B dai modelli HD è che una ripetizione estesa di poliglutammina in Htt riduce la sua connessione a PSD95, una proteina di densità postsinaptica inclusa nel clustering del recettore NMDA e kainato, causando infine una maggiore risposta di PSD95 insieme con la subunità NR2B. Recentemente, i risultati di uno studio di ricerca suggeriscono che non solo la composizione delle subunità, ma anche la localizzazione dei recettori NMDA può svolgere un ruolo fondamentale nell'attività del recettore NMDA. Un altro studio di ricerca ha dimostrato che nelle preparazioni di fette striatali gravi da topi transgenici YAC che utilizzano 128 ripetizioni CAG, i recettori NMDA extrasinaptici, in particolare quelli con NR2B, sono notevolmente aumentati rispetto ai pezzi di topi wild-type e topi YAC che esprimono Htt con 18 ripetizioni CAG. Come previsto dagli studi di ricerca in vitro, questo cambiamento è stato associato a una ridotta fosforilazione di CREB. È stato dimostrato che la maggiore percentuale di recettori NMDA extrasinaptici contenenti NR2B è associata a una maggiore localizzazione extrasinaptica di PSD95. Una via che può causare la sensibilizzazione alla stimolazione eccitotossica a valle dell'attivazione dei recettori NMDA extrasinaptici è stata identificata come attivazione di p38 MAPK. Le prove a più strati suggerite suggeriscono che la Htt mutante provoca la sensibilizzazione di MSN nell'eccitotossicità del glutammato attraverso la ridistribuzione dei recettori NMDA dalle subunità ai siti extrasinaptici.
L'attivazione dei recettori NMDA extrasinaptici nelle fettine cerebrali striatali acute può essere efficacemente mostrata nei topi YAC utilizzando 128 ripetizioni CAG attraverso lo spillover di glutammato sinaptico limitando gli EAAT. Di conseguenza, si può determinare che una ridotta espressione di EAAT può aumentare l'attivazione dei recettori NMDA. Sorprendentemente, nell'ambito dell'ibridazione situ, gli studi di ricerca hanno scoperto una diminuzione dell'espressione dell'mRNA EAAT2 astrocitico nel neostriato di tutti i pazienti con MH. Rispetto ai topi wild-type, tuttavia, non è stato riscontrato alcun cambiamento nell'espressione proteica diminuito nei sinaptosomi di topi YAC che sovraesprimono Htt umana utilizzando 128 ripetizioni CAG. I ricercatori hanno determinato che una diminuzione dell'attività EAAT2 dal modello YAC di MH era causata dalla diminuzione della palmitoilazione del trasportatore. Nei topi R6 / 2, altri hanno scoperto una diminuzione dell'mRNA EAAT2 e dell'espressione proteica associata a una diminuzione di EAAT2 nei sinaptosomi o nei pezzi cortico-striatali acuti. Tuttavia, le concentrazioni extracellulari di glutammato striatale hanno dimostrato di essere simili a quelle dei topi di controllo wild-type e una ridotta capacità di eliminazione del glutammato nei topi R6 / 2 dimostrata dalla terapia con inibitori EAAT o glutammato. Una spiegazione putativa per questo risultato potrebbe essere una diminuzione del rilascio di glutammato attraverso il sistema x? Ce in xCT, la subunità del sistema x? C che è stata dimostrata nello striato di topi R6 / 2 nei livelli di mRNA e proteine.
Come accennato in precedenza, l'iniezione del metabolita KYN QUIN in concentrazioni sovrafisiologiche è stata utilizzata come primo modello animale di MH. Ciò ha causato ulteriori studi di ricerca sul metabolismo KYN nella MH. Sorprendentemente, il precursore QUIN 3HK aggrava la neurodegenerazione dalla versione QUIN HD mentre KYNA è protettivo. Studi di ricerca hanno scoperto che nella MH allo stadio iniziale, rispetto al controllo e alla MH allo stadio terminale, le concentrazioni neostriatali di 3HK e QUIN erano notevolmente sovraregolate. Un altro studio di ricerca ha scoperto che i livelli di KYNA sono diminuiti nella MH striata sottoposta ad autopsia con il liquido cerebrospinale dei pazienti MH rispetto ai controlli. Il primo enzima di questo percorso KYN, IDO, viene attivato dallo striato di entrambi i topi YAC con 128 ripetizioni CAG. I topi carenti di IDO sono meno sensibili all'iniezione intrastriatale di QUIN. La valutazione dei metaboliti KYN da tre diversi modelli murini di topi HD, R6 / 2, YAC128 e topi knock-in HdhQ92 e HdhQ111 in varie regioni del cervello, ha suggerito l'attivazione dipendente dall'età della loro via KYN. Tuttavia, il modello dettagliato dei cambiamenti del metabolita era diverso tra le versioni con 3HK aumentato nella corteccia, nello striato e nel cervelletto nei topi R6 / 2 mentre i topi che esprimevano il mutante a grandezza naturale Htt hanno dimostrato una sovraregolazione extra corticale e striatale di QUIN. Inoltre, il trattamento di topi R6 / 2 con un inibitore KMO non permeabile alla barriera ematoencefalica, JM6, che ha indirettamente migliorato le concentrazioni di KYNA extracellulare cerebrale del 50%, è stato associato a una diminuzione dell'L-glutammato cerebrale extracellulare, diminuzione della neurodegenerazione e sopravvivenza prolungata . Ulteriori studi di ricerca sono ancora necessari per ulteriori prove.
Presi collettivamente, gli studi di ricerca supportano l'opinione che nella MH vi sia una ridistribuzione di entrambi i recettori NMDA, in particolare quelli contenenti NR2B, che possono attivare vie di segnalazione che aumentano la neurodegenerazione, come mostrato nella Figura 5. Non ci sono prove che L- cerebrale i livelli di glutammato sono notevolmente aumentati nella MH. Ciò potrebbe essere spiegato dal fatto che anche se EAAT2 e KYNA possono essere sottoregolati, c'è anche una sottoregolazione dell'azione del sistema x? C. Poiché solo livelli molto elevati di recettori NMDA attivati da QUIN, è improbabile che questo metabolita KYN contribuisca al carico eccitotossico.
In molti studi di ricerca, le prove e le misure di esito hanno dimostrato che la disregolazione del glutammato e l'eccitotossicità in molte malattie neurologiche, tra cui AD, HD e SLA, alla fine portano alla neurodegenerazione e a una varietà di sintomi associati ai problemi di salute. Lo scopo del seguente articolo è discutere e dimostrare il ruolo che la disregolazione del glutammato e l'eccitotossicità svolgono sulle malattie neurodegenerative. I meccanismi per l'eccitotossicità sono diversi per ogni problema di salute. - Dr. Alex Jimenez DC, CCST Insight - Dr. Alex Jimenez DC, CCST Insight
Modulo di valutazione metabolica
[wp-embedder-pack width = "100%" height = "1050px" download = "all" download-text = "" attachment_id = "72423 ″ /] Il seguente modulo di valutazione metabolica può essere compilato e presentato al Dr. Alex Jimenez. I gruppi di sintomi elencati in questo modulo non devono essere utilizzati come diagnosi di alcun tipo di malattia, condizione o qualsiasi altro tipo di problema di salute.
In onore del proclama del Governatore Abbott, ottobre è il mese della salute chiropratica. Impara di più riguardo la proposta.
Nell'articolo sopra, abbiamo delineato ciò che è noto sui percorsi che possono causare eccitotossicità nelle malattie neurodegenerative. Ne abbiamo anche discusso nella sclerosi laterale amiotrofica (SLA), nella malattia di Alzheimer (AD) e nella malattia di Huntington (MH) come esempi fondamentali con modelli animali sufficientemente validati negli studi di ricerca. Lo scopo delle nostre informazioni è limitato a problemi di chiropratica, salute muscoloscheletrica e nervosa, nonché articoli, argomenti e discussioni di medicina funzionale. Utilizziamo protocolli sanitari funzionali per il trattamento di lesioni o disturbi cronici del sistema muscolo-scheletrico. Per discutere ulteriormente l'argomento di cui sopra, non esitate a chiedere al Dr. Alex Jimenez o contattarci al numero 915-850-0900 .
A cura del Dr. Alex Jimenez
Riferimenti
Lewerenz, Jan e Pamela Maher. Tossicità cronica del glutammato nelle malattie neurodegenerative: quali sono le prove? Frontiere in Neuroscienze, Frontiers Media SA, 16 dic. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
Discussione argomento aggiuntiva: dolore cronico
Il dolore improvviso è una risposta naturale del sistema nervoso che aiuta a dimostrare possibili lesioni. Ad esempio, i segnali del dolore viaggiano da una regione lesa attraverso i nervi e il midollo spinale al cervello. Il dolore è generalmente meno grave poiché la lesione guarisce, tuttavia, il dolore cronico è diverso dal tipo medio di dolore. Con dolore cronico, il corpo umano continuerà a inviare segnali di dolore al cervello, indipendentemente dal fatto che la lesione sia guarita. Il dolore cronico può durare da alcune settimane a persino diversi anni. Il dolore cronico può influenzare enormemente la mobilità di un paziente e può ridurre la flessibilità, la forza e la resistenza.
Zoomer Plus neuronale per la malattia neurologica
Il dottor Alex Jimenez utilizza una serie di test per aiutare a valutare le malattie neurologiche. Lo zoom neuraleTM Plus è una gamma di autoanticorpi neurologici che offre un riconoscimento specifico anticorpo-antigene. Il vibrante Neural ZoomerTM Plus è progettato per valutare la reattività di un individuo a 48 antigeni neurologici con connessioni a una varietà di malattie neurologicamente correlate. Il vibrante zoomer neuraleTM Plus mira a ridurre le condizioni neurologiche fornendo ai pazienti e ai medici una risorsa vitale per la diagnosi precoce del rischio e una maggiore attenzione alla prevenzione primaria personalizzata.
Formule per il supporto alla metilazione
XYMOGEN s Le formule professionali esclusive sono disponibili tramite professionisti sanitari selezionati. La vendita e lo sconto su Internet delle formule XYMOGEN sono severamente vietati.
Orgogliosamente, Il dottor Alexander Jimenez rende le formule XYMOGEN disponibili solo per i pazienti sotto la nostra cura.
Si prega di chiamare il nostro ufficio in modo da poter assegnare una consulenza medica per l'accesso immediato.
Se sei un paziente di Injury Medical & Chiropractic Clinica, puoi chiedere informazioni su XYMOGEN chiamando 915-850-0900.
ï ¿½
Per vostra comodità e revisione del XYMOGEN prodotti si prega di rivedere il seguente link. *XYMOGEN-Catalog-Scaricare ï ¿½
* Tutte le precedenti politiche XYMOGEN rimangono rigorosamente in vigore.
Rispetto ad altri problemi di salute del sistema nervoso centrale (SNC), le malattie neurodegenerative croniche possono essere molto più complicate. In primo luogo, poiché la funzione mitocondriale compromessa è stata dimostrata in molte malattie neurodegenerative, i problemi risultanti nelle fonti di energia non sono così gravi come il collasso energetico nell'ictus ischemico. Pertanto, se l'eccitotossicità contribuisce alla neurodegenerazione, è necessario ipotizzare un tempo diverso di eccitotossicità cronica. Nel seguente articolo, illustreremo ciò che è noto sui percorsi che possono causare eccitotossicità nelle malattie neurodegenerative. Discuteremo specificamente che nella sclerosi laterale amiotrofica (SLA), nella malattia di Alzheimer (AD) e nella malattia di Huntington (HD) come esempi fondamentali con modelli animali sufficientemente convalidati in studi di ricerca.
Il morbo di Alzheimer
La malattia di Alzheimer (AD) è una delle principali cause di demenza tra gli anziani negli Stati Uniti. Neuropatologicamente, l'AD si caratterizza come neurodegenerazione con placche senili extracellulari costituite da? grovigli di amiloide (A?) e neurofibrillari intraneuronali di tau aggregata, che inizialmente compaiono nell'ippocampo per poi diffondersi con il progredire del problema di salute. Anche l'attivazione prominente delle cellule microgliali può essere associata all'AD. I tipi ereditari di AD si verificano a causa di mutazioni in A? proteina precursore, A? PP, o nelle preseniline, che fanno parte del complesso multiproteico coinvolto in A? generazione. La fisiopatologia dell'AD è complicata e una varietà di percorsi sono inclusi nella degenerazione sinaptica e cellulare nell'AD, come anomalie nelle vie di segnalazione attraverso la glicogeno sintasi chinasi-3 beta o proteine chinasi attivate dal mitogeno, rientro del ciclo cellulare, ossidativo stress o diminuzione del trasporto di fattori trofici e disregolazione surrenale. Tuttavia, le prove suggeriscono che la disregolazione dell'L-glutammato gioca un ruolo critico nella malattia di Alzheimer.
Studi di ricerca hanno dimostrato che i neuroni primari di topi transgenici che sovraesprimono la presenilina mutante sono molto più sensibili alla stimolazione eccitotossica in vitro. In vitro, aggregato A? aumenta la tossicità del L-glutammato mediata dal recettore NMDA e kainato, forse interrompendo l'omeostasi del calcio neuronale. Altri hanno dimostrato che A? può aumentare l'eccitabilità neuronale modificando la capacità della glicogeno sintasi chinasi 3? inibizione per diminuire le vie mediate dal recettore NMDA. Solubile A? È stato dimostrato che gli oligomeri causano il rilascio di L-glutammato dagli astrociti con conseguente perdita della colonna vertebrale dendritica attraverso l'eccessiva attivazione dei recettori NMDA extrasinaptici. Inoltre, è stato dimostrato che le concentrazioni extracellulari di L-glutammato aumentano in un modello di topo transgenico triplo di AD, in cui un trattamento di 3 mesi con l'inibitore del recettore NMDA alla fine ha influenzato la perdita di sinapsi. Tuttavia, sono ancora necessari ulteriori studi di ricerca.
Numerosi studi di ricerca sui topi hanno dimostrato le conseguenze della patologia simile all'AD sull'espressione e / o sulla funzione di EAAT. Nelle preparazioni a fette acute dell'ippocampo, A? è stato dimostrato che interrompe la clearance dell'L-glutammato rilasciato sinapticamente diminuendo l'inserimento nella membrana di EAAT2, un risultato forse mediato dallo stress ossidativo. In topi A? PP23 di età, studi di ricerca hanno rivelato la sottoregolazione dell'espressione di EAAT2 nella corteccia frontale e nell'ippocampo, che nella corteccia frontale era associata ad un aumento dell'espressione di xCT. Questi cambiamenti sono stati associati a una forte tendenza a migliorare la quantità di L-glutammato extracellulare misurata mediante microdialisi. In topi AD transgenici tripli che esprimono le mutazioni della proteina precursore dell'amiloide K670N e M671L, la mutazione M1V della presenilina 146 e la mutazione tau P301L, è stata dimostrata una forte diminuzione dell'espressione di EAAT2 dipendente dall'età. Il ripristino dell'attività EAAT2 nei topi AD in seguito al trattamento con tutto l'antibiotico α-lattamico Cef è stato associato a una diminuzione del deterioramento cognitivo e alla riduzione della patologia tau. Nel cervello umano di AD, è stata determinata una diminuzione dell'espressione della proteina EAAT2 e una diminuzione dell'azione EAAT. Tuttavia, questa misura di risultato non può essere replicata da altri ricercatori. A livello di trascrittoma, studi di ricerca hanno scoperto variazioni di giunzione a salto dell'esone di EAAT2 che riducono l'attività di trasporto del glutammato per essere sovraregolate nel cervello umano di AD. Dal CSF, diversi gruppi hanno dimostrato un aumento delle concentrazioni di glutammato nei pazienti con AD in cui altri gruppi hanno dimostrato assolutamente nessun cambiamento o addirittura una diminuzione dei livelli di L-glutammato associati alla malattia di Alzheimer.
In vitro, A? provoca la secrezione di L-glutammato dalla microglia primaria attraverso la sovraregolazione del programma x? c. Altri hanno scoperto che ha anche innescato il rilascio di L-glutammato dagli astrociti attraverso l'attivazione del recettore nicotinico dell'acetilcolina? 7. Inoltre, xCT, la subunità specifica del sistema x? C è sovraregolata nella regione delle placche senili, possibilmente nelle cellule microgliali, nei topi Thy1-APP751 (TgAPP) che esprimono APP umana recanti lo svedese (S: KM595 / 596NL) e Londra ( L: V6421) mutazioni dopo A? iniezione nell'ippocampo. Le valutazioni immunoblot semiquantitative hanno rivelato una sovraregolazione dell'espressione della proteina xCT nella corteccia frontale nei topi anziani A? PP23 rispetto ai controlli wild-type.
Studi di ricerca post-mortem mostrano che il metabolismo di KYN influisce su concentrazioni elevate di KYNA da AD mentre viene scoperto anche nei gangli della base di entrambi i malati di AD. Utilizzando l'immunoistochimica, studi di ricerca hanno dimostrato che l'immunoreattività sia per IDO che per QUIN sovraregolata nei cervelli di AD, in particolare in prossimità delle placche. UN? provoca l'espressione di IDO nei macrofagi primari umani e nella microglia. L'inibizione sistemica di KMO alla fine aumenta i livelli di KYNA cerebrale e ha migliorato il fenotipo di un modello murino di AD, indicando che una sovraregolazione di KYNA può essere una reazione protettiva endogena, tra cui l'inibitore IDO, coptisina, diminuzione della microglia, attivazione astrocitica e deterioramento cognitivo nei topi AD .
Presi insieme, insieme a molti altri cambiamenti dannosi, ci sono prove di eccitotossicità cronica nell'AD che può essere guidata da numerose variabili, tra cui la sensibilizzazione centrale di entrambi i recettori NMDA, una diminuzione della capacità di ricaptazione di L-glutammato e L-aspartato e un aumento nel rilascio di glutammato attraverso il sistema x? c, come mostrato nella Figura 4. Sebbene la via KYN sembri essere sovraregolata nell'AD, non è possibile trarre conclusioni specifiche sulla neurotrasmissione glutamatergica dalla sovraregolazione dei due QUIN che era neurotossico e neuroprotettivo KYNA.
ï ¿½
In molti studi di ricerca, le prove e le misure di esito hanno dimostrato che la disregolazione del glutammato e l'eccitotossicità in molte malattie neurologiche, tra cui AD, HD e SLA, alla fine portano alla neurodegenerazione e a una varietà di sintomi associati ai problemi di salute. Lo scopo del seguente articolo è discutere e dimostrare il ruolo che la disregolazione del glutammato e l'eccitotossicità svolgono sulle malattie neurodegenerative. I meccanismi per l'eccitotossicità sono diversi per ogni problema di salute. - Dr. Alex Jimenez DC, CCST Insight - Dr. Alex Jimenez DC, CCST Insight
Nell'articolo sopra, abbiamo delineato ciò che è noto sui percorsi che possono causare eccitotossicità nelle malattie neurodegenerative. Ne abbiamo anche discusso nella sclerosi laterale amiotrofica (SLA), nella malattia di Alzheimer (AD) e nella malattia di Huntington (MH) come esempi fondamentali con modelli animali sufficientemente validati negli studi di ricerca. Lo scopo delle nostre informazioni è limitato a problemi di chiropratica, salute muscoloscheletrica e nervosa, nonché articoli, argomenti e discussioni di medicina funzionale. Utilizziamo protocolli sanitari funzionali per il trattamento di lesioni o disturbi cronici del sistema muscolo-scheletrico. Per discutere ulteriormente l'argomento di cui sopra, non esitate a chiedere al Dr. Alex Jimenez o contattarci al numero 915-850-0900 .
A cura del Dr. Alex Jimenez
Riferimenti ï ¿½
Lewerenz, Jan e Pamela Maher. Tossicità cronica del glutammato nelle malattie neurodegenerative: quali sono le prove? Frontiere in Neuroscienze, Frontiers Media SA, 16 dic. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
Discussione argomento aggiuntiva: dolore cronico
Il dolore improvviso è una risposta naturale del sistema nervoso che aiuta a dimostrare possibili lesioni. Ad esempio, i segnali del dolore viaggiano da una regione lesa attraverso i nervi e il midollo spinale al cervello. Il dolore è generalmente meno grave poiché la lesione guarisce, tuttavia, il dolore cronico è diverso dal tipo medio di dolore. Con dolore cronico, il corpo umano continuerà a inviare segnali di dolore al cervello, indipendentemente dal fatto che la lesione sia guarita. Il dolore cronico può durare da alcune settimane a persino diversi anni. Il dolore cronico può influenzare enormemente la mobilità di un paziente e può ridurre la flessibilità, la forza e la resistenza.
Zoomer Plus neuronale per la malattia neurologica
Il dottor Alex Jimenez utilizza una serie di test per aiutare a valutare le malattie neurologiche. Lo zoom neuraleTM Plus è una gamma di autoanticorpi neurologici che offre un riconoscimento specifico anticorpo-antigene. Il vibrante Neural ZoomerTM Plus è progettato per valutare la reattività di un individuo a 48 antigeni neurologici con connessioni a una varietà di malattie neurologicamente correlate. Il vibrante zoomer neuraleTM Plus mira a ridurre le condizioni neurologiche fornendo ai pazienti e ai medici una risorsa vitale per la diagnosi precoce del rischio e una maggiore attenzione alla prevenzione primaria personalizzata.
Formule per il supporto alla metilazione
XYMOGEN s Le formule professionali esclusive sono disponibili tramite professionisti sanitari selezionati. La vendita e lo sconto su Internet delle formule XYMOGEN sono severamente vietati.
Orgogliosamente, Il dottor Alexander Jimenez rende le formule XYMOGEN disponibili solo per i pazienti sotto la nostra cura.
Si prega di chiamare il nostro ufficio in modo da poter assegnare una consulenza medica per l'accesso immediato.
Se sei un paziente di Injury Medical & Chiropractic Clinica, puoi chiedere informazioni su XYMOGEN chiamando 915-850-0900.
ï ¿½
Per vostra comodità e revisione del XYMOGEN prodotti si prega di rivedere il seguente link. *XYMOGEN-Catalog-Scaricare ï ¿½
* Tutte le precedenti politiche XYMOGEN rimangono rigorosamente in vigore.
Rispetto ad altri problemi di salute del sistema nervoso centrale (SNC), le malattie neurodegenerative croniche possono essere molto più complicate. In primo luogo, poiché la funzione mitocondriale compromessa è stata dimostrata in molte malattie neurodegenerative, i problemi risultanti nelle fonti di energia non sono così gravi come il collasso energetico nell'ictus ischemico. Pertanto, se l'eccitotossicità contribuisce alla neurodegenerazione, è necessario ipotizzare un tempo diverso di eccitotossicità cronica. Nel seguente articolo, illustreremo ciò che è noto sui percorsi che possono causare eccitotossicità nelle malattie neurodegenerative. Discuteremo specificamente che nella sclerosi laterale amiotrofica (SLA), nella malattia di Alzheimer (AD) e nella malattia di Huntington (HD) come esempi fondamentali con modelli animali sufficientemente convalidati in studi di ricerca.
Sclerosi laterale amiotrofica
La sclerosi laterale amiotrofica (SLA) è una malattia neurodegenerativa associata alla degenerazione dei motoneuroni che alla fine determinano la durata del problema di salute. La SLA è considerata fatale diversi anni dopo l'inizio. Si ipotizza che l'eccitotossicità dell'L-glutammato giochi un ruolo nella morte dei motoneuroni nella SLA perché le cellule mostrano livelli aumentati di recettori AMPA permeabili al calcio e bassi livelli di proteine leganti il calcio. Rispetto all'utilizzo di AMPA e kainato e L-HCA, nel midollo spinale dei ratti, il trattamento con neuroni motori risparmiati dall'NMDA suggerisce che l'eccitotossicità dell'NMDA potrebbe in realtà non svolgere un ruolo fondamentale nella SLA. Tuttavia, l'eccitotossicità mediata dal recettore NMDA nei motoneuroni è stata dimostrata in colture di fettine organotipiche di embrioni di pollo. Studi di ricerca elettrofisiologica hanno suggerito che l'ipereccitabilità transitoria dei nervi motori nella fase presintomatica della SLA nei topi transgenici per la mutazione G93A della SOD1 umana è associata alla SLA ereditaria. Inoltre, l'ipereccitabilità corticale è stata registrata nei pazienti affetti da SLA familiare e sporadica con l'insorgenza di sintomi nei portatori di mutazione familiare della SLA. Inoltre, l'unico farmaco approvato e / o farmaco utilizzato per la SLA, che aumenta la sopravvivenza da 2 a 3 mesi, agisce come un inibitore dei recettori NMDA e kainato insieme alla rapida sovraregolazione dell'attività EAAT nei sinaptosomi, secondo diversi studi di ricerca.
Nel midollo spinale sottoposto ad autopsia da pazienti con SLA, diversi gruppi hanno dimostrato una diminuzione dell'espressione della proteina EAAT2 e non dell'espressione della proteina EAAT1 nella materia grigia delle regioni con considerevole perdita di motoneuroni. Inoltre, sia l'assorbimento di L-glutammato che l'immunoreattività EAAT2, come dimostrato dal Western blotting, hanno dimostrato di essere quantitativamente diminuite nel tessuto post-mortem dei pazienti con SLA, in particolare nel midollo spinale, il tessuto più comunemente colpito dal problema di salute. Inoltre, è stato dimostrato che come possibile effetto della sottoregolazione EAAT2, le quantità di L-glutammato sono aumentate nel liquido cerebrospinale nei pazienti con SLA. Tuttavia, questa misura di risultato non può essere replicata da altri studi di ricerca.
La sottoregolazione di EAAT2 nella SLA umana è dimostrata in diversi modelli animali di SLA, inclusi topi transgenici che esprimono SOD1 umana contenente la mutazione G93A che causa la SLA ereditaria o ratti transgenici che esprimono la stessa mutazione. Sorprendentemente, "mentre Bendotti ha dimostrato una diminuzione tardiva nell'espressione di EAAT2 nel momento in cui i topi erano già diventati sintomatici", studi di ricerca hanno dimostrato fluttuazioni nell'espressione di EAAT2 allo stadio presintomatico. L'antibiotico α-lattamico ceftriaxone (Cef) promuove la produzione di EAAT2 in fettine di midollo spinale murino in coltura e in co-colture neurone / astrocita. Inoltre, ha causato l'espressione di EAAT2 dal midollo spinale di topi G93A mSOD1 Tg wild-type e mutanti, che è stata associata a una diminuzione della perdita dei motoneuroni, riduzione del peso e altri sintomi simili alla SLA, nonché un aumento della sopravvivenza , compatibile con l'ipotesi che la perdita di EAAT2 contribuisca all'eccitotossicità cronica in questo modello murino. Solo di recente, una diminuzione significativa dell'immunoreattività EAAT2 è stata dimostrata in un modello di corteccia separato per la SLA, ratti che esprimono la proteina di legame del DNA TAR mutante 43 che induce la SLA solo negli astrociti. Sorprendentemente, gli studi di ricerca hanno dimostrato che, misurate mediante microdialisi, le concentrazioni extracellulari di L-glutammato e L-aspartato aumentano mentre la capacità di eliminazione dell'L-glutammato diminuisce nella corteccia cerebrale dei topi G93A mSOD1 Tg, tuttavia, questa regione non mostra evidente patologia né sottoregolazione di EAAT1 quando valutati.
Presi insieme questi studi di ricerca supportano l'opinione che vi sia una sottoregolazione di EAAT2 sia nei pazienti con SLA umana che nei modelli animali di SLA. Tuttavia, mentre alcuni studi di ricerca sugli animali suggeriscono che la sottoregolazione EAAT2 si verifica prima della perdita dei motoneuroni, altri sono compatibili con l'ipotesi che la sottoregolazione di EAAT2, la cui espressione astrogliale è associata all'esistenza di neuroni, sia una conseguenza della neurodegenerazione nelle malattie neurologiche. .
Inoltre, gli EAAT riducono l'L-glutammato extracellulare, l'L-glutammato cerebrale extracellulare è sovraregolato in una varietà di regioni del cervello dal sistema antiportatore di cistina / glutammato x? C. XCT, una particolare subunità del programma x? C, ha dimostrato di essere regolata e mantenuta in modo differenziale nei modelli murini di SLA. Studi di ricerca hanno dimostrato che l'assorbimento di cistina radiomarcata era sovraregolato in fettine di midollo spinale di topi presintomatici G93A mSOD1 Tg all'età di 70 giorni ma non in 55 o 100 giorni e non in topi sintomatici di 130 giorni, il che ha anche determinato che la sovraregolazione di l'assorbimento della cistina al giorno 70 era dovuto all'attività del sistema x? c che utilizzava l'inibitore del sistema x? c sulfasalazina (SSZ). Va considerato, tuttavia, che la cistina può anche essere trasportata dagli EAAT. Pertanto, poiché le prove sulla sensibilità alla SSZ dell'assunzione di cistina non sono state dimostrate per i giorni 100 e 130, l'assorbimento differenziale di cistina dimostrato in questo studio di ricerca nelle età più anziane potrebbe piuttosto essere il risultato di una ridotta azione EAAT. In confronto, studi di ricerca con rtPCR hanno dimostrato una forte crescita dei livelli di mRNA di xCT nei topi G37R mSOD1 Tg all'inizio dei sintomi, che è stata ulteriormente aumentata con il miglioramento dei sintomi. Inoltre, è stato dimostrato che xCT è stato dimostrato principalmente nelle cellule microgliali del midollo spinale. Microglia ha rivelato la sovraregolazione dell'mRNA di xCT nella fase presintomatica. Prese insieme, queste misure di risultato suggeriscono che il sistema x? C è sovraregolato nei modelli animali di SLA. Tuttavia, mancano le prove sul fatto che questo sia vero per i casi umani di SLA. Tuttavia, ulteriori studi di ricerca hanno rivelato che i livelli di mRNA di CD68, un marker di attivazione della microglia, erano associati all'espressione di mRNA di xCT nel tessuto del midollo spinale post mortem di individui con SLA, dimostrando che la neuroinfiammazione negli esseri umani è anche associata alla sovraregolazione xCT.
Oltre alla disregolazione dei livelli di L-glutammato e L-aspartato da parte della sottoregolazione EAAT o della sovraregolazione del sistema x? C, è stato suggerito che anche i percorsi che regolano e mantengono indirettamente la neurotrasmissione glutamatergica partecipino alla degenerazione dei motoneuroni nella SLA. È stato dimostrato che i livelli di D-serina aumentano notevolmente dal midollo spinale dei topi G93A mSOD1 Tg. Partendo dall'esordio della malattia e continuando nel corso di questa fase sintomatica, la D-serina aumenta l'eccitotossicità dell'NMDA nei motoneuroni. La sovraregolazione della D-serina nel midollo spinale è stata duplicata da altri studi di ricerca. La sottoregolazione di questo enzima DAO che metabolizza la D-serina nel tratto reticolospinale è stata dimostrata come il meccanismo principale per la sovraregolazione della D-serina nel midollo spinale nei topi SLA. Inoltre, l'inattivazione genetica di DAO nei topi è stata associata alla degenerazione dei motoneuroni e una carenza dell'enzima D-serina che genera la serina racemasi ha prolungato la sopravvivenza nei topi G93A mSOD1 Tg sebbene abbia accelerato l'insorgenza della malattia neurodegenerativa. È stato dimostrato che una mutazione eterozigote di DAO è separata dal fenotipo SLA in una famiglia numerosa con SLA ereditaria. Tuttavia, questa continua ad essere l'unica famiglia determinata in cui una mutazione DAO è associata alla SLA.
Per quanto riguarda l'altro amminoacido co-agonista del recettore NMDA, la glicina, un aumento dei livelli di liquido cerebrospinale nei pazienti con SLA è stato dimostrato da un gruppo, tuttavia, non può essere replicato da altri studi di ricerca. Diversi studi di ricerca hanno anche determinato che i livelli di KYNA sono sovraregolati nel liquido cerebrospinale dei pazienti con SLA bulbare e in quelli in stadio terminale di SLA. Indipendentemente, è stato rivelato che i livelli di triptofano e KYN sono aumentati nel liquido cerebrospinale dei pazienti con SLA rispetto ai controlli. Inoltre, è stato dimostrato che IDO è espresso nei neuroni e nella microglia del midollo spinale di pazienti con SLA, indicando che l'attivazione microglia può aumentare, tra gli altri, la conversione del triptofano nella SLA in KYN.
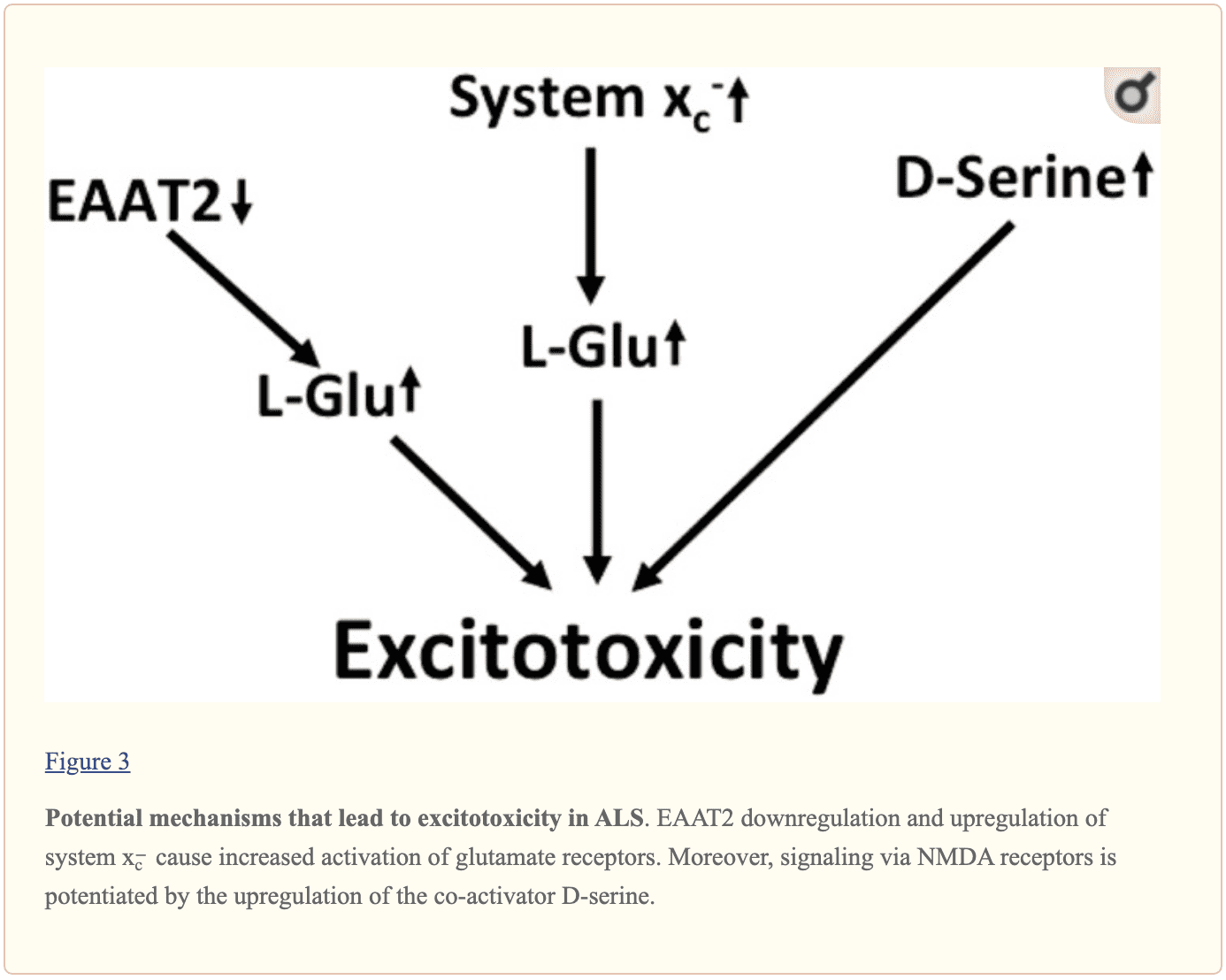
Prove multistrato suggeriscono che l'aumento della neurotrasmissione glutamatergica è all'interno della SLA e può alla fine causare neurodegenerazione nelle malattie neurodegenerative, come mostrato nella Figura 3. La sottoregolazione di EAAT2 negli astrociti e la sovraregolazione del programma x? Previsto nel contesto dell'attivazione della microglia sono state ripetutamente documentate. Anche i recettori NMDA della D-serina possono svolgere un ruolo nella disregolazione. Inoltre, la via della chinurenina sembra essere attivata nella SLA.
ï ¿½
In molti studi di ricerca, prove e misure di esito hanno dimostrato che l'eccitotossicità cronica può essere associata a una varietà di malattie neurodegenerative, tra cui AD, HD e SLA, causando infine neurodegenerazione e una varietà di sintomi associati ai problemi di salute. Lo scopo del seguente articolo è di delineare ciò che può causare eccitotossicità nelle malattie neurodegenerative. Ne discuteremo nella sclerosi laterale amiotrofica (SLA), nella malattia di Alzheimer (AD) e nella malattia di Huntington (MH). - Dr. Alex Jimenez DC, CCST Insight - Dr. Alex Jimenez DC, CCST Insight
Nell'articolo sopra, abbiamo delineato ciò che è noto sui percorsi che possono causare eccitotossicità nelle malattie neurodegenerative. Ne abbiamo anche discusso nella sclerosi laterale amiotrofica (SLA), nella malattia di Alzheimer (AD) e nella malattia di Huntington (MH) come esempi fondamentali con modelli animali sufficientemente validati negli studi di ricerca. Lo scopo delle nostre informazioni è limitato a problemi di chiropratica, salute muscoloscheletrica e nervosa, nonché articoli, argomenti e discussioni di medicina funzionale. Utilizziamo protocolli sanitari funzionali per il trattamento di lesioni o disturbi cronici del sistema muscolo-scheletrico. Per discutere ulteriormente l'argomento di cui sopra, non esitate a chiedere al Dr. Alex Jimenez o contattarci al numero 915-850-0900 .
A cura del Dr. Alex Jimenez
Riferimenti ï ¿½
Lewerenz, Jan e Pamela Maher. Tossicità cronica del glutammato nelle malattie neurodegenerative: quali sono le prove? Frontiere in Neuroscienze, Frontiers Media SA, 16 dic. 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4679930/.
Discussione argomento aggiuntiva: dolore cronico
Il dolore improvviso è una risposta naturale del sistema nervoso che aiuta a dimostrare possibili lesioni. Ad esempio, i segnali del dolore viaggiano da una regione lesa attraverso i nervi e il midollo spinale al cervello. Il dolore è generalmente meno grave poiché la lesione guarisce, tuttavia, il dolore cronico è diverso dal tipo medio di dolore. Con dolore cronico, il corpo umano continuerà a inviare segnali di dolore al cervello, indipendentemente dal fatto che la lesione sia guarita. Il dolore cronico può durare da alcune settimane a persino diversi anni. Il dolore cronico può influenzare enormemente la mobilità di un paziente e può ridurre la flessibilità, la forza e la resistenza.
Zoomer Plus neuronale per la malattia neurologica
ï ¿½
Il dottor Alex Jimenez utilizza una serie di test per aiutare a valutare le malattie neurologiche. Lo zoom neuraleTM Plus è una gamma di autoanticorpi neurologici che offre un riconoscimento specifico anticorpo-antigene. Il vibrante Neural ZoomerTM Plus è progettato per valutare la reattività di un individuo a 48 antigeni neurologici con connessioni a una varietà di malattie neurologicamente correlate. Il vibrante zoomer neuraleTM Plus mira a ridurre le condizioni neurologiche fornendo ai pazienti e ai medici una risorsa vitale per la diagnosi precoce del rischio e una maggiore attenzione alla prevenzione primaria personalizzata.
Formule per il supporto alla metilazione
XYMOGEN s Le formule professionali esclusive sono disponibili tramite professionisti sanitari selezionati. La vendita e lo sconto su Internet delle formule XYMOGEN sono severamente vietati.
Orgogliosamente, Il dottor Alexander Jimenez rende le formule XYMOGEN disponibili solo per i pazienti sotto la nostra cura.
Si prega di chiamare il nostro ufficio in modo da poter assegnare una consulenza medica per l'accesso immediato.
Se sei un paziente di Injury Medical & Chiropractic Clinica, puoi chiedere informazioni su XYMOGEN chiamando 915-850-0900.
ï ¿½
Per vostra comodità e revisione del XYMOGEN prodotti si prega di rivedere il seguente link. *XYMOGEN-Catalog-Scaricare ï ¿½
* Tutte le precedenti politiche XYMOGEN rimangono rigorosamente in vigore.
Lo strumento Find A Practitioner di IFM è la più grande rete di riferimento in Medicina Funzionale, creata per aiutare i pazienti a localizzare professionisti di Medicina Funzionale in qualsiasi parte del mondo. I professionisti certificati IFM sono elencati per primi nei risultati di ricerca, data la loro vasta formazione in Medicina Funzionale
Prenotazione online e appuntamenti 24 ore su 7, XNUMX giorni su XNUMX *





 ï ¿½
ï ¿½ 
 ï ¿½
ï ¿½




