Back Clinic Team di Chiropratica e Medicina Funzionale Stress Ossidativo. Lo stress ossidativo è definito come un disturbo nell'equilibrio tra la produzione di ossigeno reattivo (radicali liberi) e le difese antiossidanti. In altre parole, si tratta di uno squilibrio tra la produzione di radicali liberi e la capacità dell'organismo di contrastare o disintossicare gli effetti nocivi attraverso la neutralizzazione da parte degli antiossidanti. Lo stress ossidativo porta a molte condizioni fisiopatologiche nel corpo. Questi includono malattie neurodegenerative, ad esempio morbo di Parkinson, morbo di Alzheimer, mutazioni genetiche, tumori, sindrome da stanchezza cronica, sindrome dell'X fragile, disturbi cardiaci e dei vasi sanguigni, aterosclerosi, insufficienza cardiaca, infarto e malattie infiammatorie. L'ossidazione avviene in una serie di circostanze:
le cellule usano il glucosio per produrre energia
il sistema immunitario sta combattendo contro i batteri e creando infiammazioni
i corpi detossificano inquinanti, pesticidi e fumo di sigaretta
Ci sono milioni di processi che avvengono nei nostri corpi in un dato momento che possono provocare l'ossidazione. Ecco alcuni sintomi:
stanchezza
Perdita di memoria e / o nebbia cerebrale
Dolore muscolare e articolare
Rughe e capelli grigi
Diminuzione della vista
Mal di testa e sensibilità al rumore
Suscettibilità alle infezioni
Scegliere cibi biologici ed evitare le tossine nel proprio ambiente fa una grande differenza. Questo, insieme alla riduzione dello stress, può essere utile per ridurre l'ossidazione.
Per le persone che desiderano migliorare la salute del cuore, il consumo di prugne può aiutare a sostenere la salute cardiovascolare?
Prugne e salute del cuore
Le prugne, o prugne secche, sono frutti ricchi di fibre che sono più nutrienti delle prugne fresche e aiutano la digestione e il movimento intestinale. (Ellen Lever et al., 2019) Una nuova ricerca suggerisce che potrebbero offrire qualcosa di più del semplice sollievo dalla digestione e dalla stitichezza, secondo nuovi studi presentati all'American Society for Nutrition. Mangiare prugne ogni giorno può migliorare i livelli di colesterolo e ridurre lo stress ossidativo e l’infiammazione.
Mangiare da cinque a dieci prugne al giorno può favorire la salute del cuore.
Negli uomini sono stati osservati benefici per la salute del cuore derivanti dal consumo regolare.
Nelle donne anziane, il consumo regolare di prugne secche non ha avuto effetti negativi sui livelli di colesterolo totale, zucchero nel sangue e insulina.
Un altro studio ha scoperto che mangiare 50-100 grammi o da cinque a dieci prugne al giorno era associato a una riduzione del rischio di malattie cardiache. (Mee Young Hong et al., 2021)
Le riduzioni del colesterolo e dei marcatori di infiammazione sono dovute al miglioramento dei livelli di antiossidanti.
La conclusione è stata che le prugne possono supportare la salute cardiovascolare.
Prugne e prugne fresche
Sebbene gli studi abbiano suggerito che le prugne possano favorire la salute del cuore, ciò non significa che le prugne fresche o il succo di prugna possano offrire gli stessi benefici. Tuttavia, non ci sono molti studi sui benefici delle prugne fresche o del succo di prugna, ma è possibile che lo siano. Tuttavia, sono necessarie ulteriori ricerche. Le prugne fresche essiccate all'aria calda migliorano il valore nutrizionale e la durata di conservazione del frutto, il che potrebbe essere il motivo per cui la versione essiccata conserva più sostanze nutritive. (Harjeet Singh Brar e altri, 2020)
Gli individui potrebbero dover mangiare più prugne per ottenere gli stessi benefici.
Mangiare 5-10 prugne sembra essere più facile che cercare di eguagliare la stessa quantità, o più, di prugne fresche.
Ma entrambe le opzioni sono consigliate al posto del succo di prugna poiché i frutti interi contengono più fibre, fanno sentire il corpo più pieno e hanno meno calorie.
Vantaggi per i giovani
La maggior parte della ricerca è stata condotta su donne in postmenopausa e uomini sopra i 55 anni, ma anche i soggetti più giovani possono trarre beneficio dal consumo di prugne secche. Una dieta ricca di frutta e verdura è considerata sana, quindi aggiungere le prugne alla propria dieta aumenterà i benefici per la salute. Per le persone a cui non piacciono le prugne, anche frutti come mele e frutti di bosco sono consigliati per la salute del cuore. Tuttavia, la frutta costituisce solo una parte della dieta ed è importante concentrarsi su una dieta equilibrata con verdure, legumi e oli salutari per il cuore. Le prugne contengono molte fibre, quindi si consiglia agli individui di aggiungerle lentamente nella loro routine quotidiana, poiché aggiungerne troppe in una volta può portare a crampi, gonfiore e/o costipazione.
Superare l’insufficienza cardiaca congestizia
Riferimenti
Lever, E., Scott, S. M., Louis, P., Emery, P. W. e Whelan, K. (2019). L’effetto delle prugne sulla produzione di feci, sul tempo di transito intestinale e sul microbiota gastrointestinale: uno studio randomizzato e controllato. Nutrizione clinica (Edimburgo, Scozia), 38(1), 165–173. doi.org/10.1016/j.clnu.2018.01.003
Hong, M. Y., Kern, M., Nakamichi-Lee, M., Abbaspour, N., Ahouraei Far, A., & Hooshmand, S. (2021). Il consumo di prugne secche migliora il colesterolo totale e la capacità antiossidante e riduce l'infiammazione nelle donne sane in postmenopausa. Giornale di alimenti medicinali, 24(11), 1161–1168. doi.org/10.1089/jmf.2020.0142
Harjeet Singh Brar, Prabhjot Kaur, Jayasankar Subramanian, Gopu R. Nair e Ashutosh Singh (2020) Effetto del pretrattamento chimico sulla cinetica dell'essiccazione e sulle caratteristiche fisico-chimiche delle prugne gialle europee, International Journal of Fruit Science, 20:sup2, S252-S279 , DOI: 10.1080/15538362.2020.1717403
Dr. Alex Jimenez, DC, presenta come lo stress cronico può avere un impatto sul corpo e come è correlato con l'infiammazione in questa serie in 2 parti. Parte 1 ha esaminato in che modo lo stress è correlato a vari sintomi che influenzano i livelli genici del corpo. La parte 2 esamina come l'infiammazione e lo stress cronico sono correlati ai vari fattori che possono portare allo sviluppo fisico. Indirizziamo i nostri pazienti a fornitori di servizi medici certificati che forniscono trattamenti disponibili per molte persone che soffrono di stress cronico associato al sistema cardiovascolare, endocrino e immunitario che colpisce il corpo e sviluppa infiammazioni. Incoraggiamo ciascuno dei nostri pazienti menzionandoli ai fornitori di servizi medici associati in base alla loro analisi in modo appropriato. Comprendiamo che l'educazione è un modo delizioso per porre domande ai nostri fornitori su richiesta e comprensione del paziente. Il dottor Jimenez, DC, utilizza queste informazioni solo come servizio educativo. Negazione di responsabilità
In che modo lo stress può influire su di noi?
Il dottor Alex Jimenez, DC, presenta: Lo stress può creare molte emozioni che possono avere un impatto enorme su molti di noi. Che si tratti di rabbia, frustrazione o tristezza, lo stress può portare chiunque a raggiungere un punto di rottura e causare condizioni sottostanti che possono trasformarsi in problemi cardiovascolari. Quindi quelle persone con il più alto livello di rabbia, quando guardi la letteratura cardiovascolare, hanno la minima probabilità di sopravvivenza. La rabbia è un cattivo giocatore. La rabbia provoca aritmia. Questo studio ha esaminato, ora che abbiamo persone con ICD e defibrillatori, possiamo monitorare queste cose. E vediamo che la rabbia può scatenare aritmie ventricolari nei pazienti. Ed ora è facile da seguire, con parte della nostra tecnologia.
La rabbia è stata collegata a episodi di fibrillazione atriale. Quando ci pensi, è l'adrenalina che si riversa nel corpo e provoca la costrizione coronarica. Sta aumentando la frequenza cardiaca. Tutte queste cose possono portare all'aritmia. E non deve essere fibrillazione atriale. Può essere APC e VPC. Ora, sono emerse alcune ricerche molto interessanti sulla telomerasi e sui telomeri. I telomeri sono piccoli cappucci sui cromosomi e la telomerasi è l'enzima legato alla formazione dei telomeri. E ora, possiamo capire attraverso il linguaggio della scienza, e stiamo iniziando a usare la tecnologia e usare la scienza in un modo che non avremmo mai potuto fare prima per capire l'impatto dello stress sui telomeri e sugli enzimi telomerasi.
I fattori che portano allo stress cronico
Il dottor Alex Jimenez, DC, presenta: Quindi una delle persone chiave per studiare questo è la vincitrice del premio Nobel, la dottoressa Elizabeth Blackburn. E quello che ha detto è che questa è una conclusione, e torneremo ad alcuni dei suoi altri studi. Ci dice che i telomeri dei bambini delle donne in utero avevano molto stress o erano ancora più corti nella giovane età adulta rispetto alle madri che non avevano le stesse situazioni stressanti. Lo stress psicologico materno durante la gravidanza può esercitare un effetto di programmazione sullo sviluppo del sistema biologico dei telomeri che è già evidente alla nascita, come si evince dall'impostazione della lunghezza telemetrica dei leucociti appena nati. Quindi i bambini possono entrare impressi e, anche se lo fanno, questo può essere trasformato.
Che dire della discriminazione razziale, queste scatole qui mostrano un'elevata discriminazione razziale che porta a una bassa lunghezza dei telomeri, a cui la maggior parte di noi ha mai pensato. Quindi, la lunghezza più corta dei telomeri porta ad un aumento del rischio di cancro e mortalità generale. I tassi di incidenza del cancro sono 22.5 per 1000 anni-persona nel gruppo con telomeri più corti, contro 14.2 nel gruppo medio e 5.1 nel gruppo con telomeri più lunghi. Telomeri più corti possono portare all'instabilità del cromosoma e provocare la formazione del cancro. Quindi, ora comprendiamo, attraverso il linguaggio della scienza, l'impatto dello stress sull'enzima telomerasi e sulla lunghezza dei telomeri. Secondo la dottoressa Elizabeth Blackburn, 58 donne in premenopausa si prendevano cura dei loro figli malati cronici rispetto alle donne che avevano figli sani. Alle donne è stato chiesto come percepiscono lo stress nella loro vita e se influisce sulla loro salute influenzando il loro invecchiamento cellulare.
Questa era la domanda dello studio mentre osservavano la lunghezza dei telomeri e l'enzima telomerasi, e questo è ciò che hanno scoperto. Ora, la parola chiave qui è percepita. Non dobbiamo giudicare lo stress dell'altro. Lo stress è personale e alcune delle nostre risposte potrebbero essere genetiche. Ad esempio, qualcuno che ha comp omozigoti con un gene pigro può avere molta più ansia di qualcuno che non ha questo polimorfismo genetico. Qualcuno che ha un MAOA in un MAOB può avere più ansia di qualcuno che non ha quel polimorfismo genetico. Quindi c'è una componente genetica nella nostra risposta, ma quello che ha scoperto è stato lo stress psicologico percepito. E il numero di anni di cura dei bambini con malattie croniche è stato associato a una minore lunghezza dei telomeri e a una minore attività della telomerasi, fornendo la prima indicazione che lo stress può influire sul mantenimento e sulla longevità dei telomeri.
Come trasformare la nostra risposta allo stress?
Il dottor Alex Jimenez, DC, presenta: È potente e molti operatori sanitari sono sotto qualche forma di stress. E la domanda è: cosa possiamo fare per trasformare la nostra risposta? Framingham ha anche esaminato la depressione e ha identificato la depressione clinica come un rischio maggiore di eventi cardiovascolari e scarsi risultati rispetto a fumo, diabete, LDL alto e HDL basso, il che è pazzesco perché passiamo tutto il nostro tempo su queste cose. Tuttavia, non dedichiamo molto tempo ad affrontare gli aspetti emotivi delle malattie vascolari. Questo è depressione influenzata, inventario, un semplice test di screening per la depressione, guardando le persone con alti livelli di depressione rispetto a bassi livelli di depressione. E puoi vedere che mentre passi dal livello più basso a quello più alto, mentre ti fai strada, le possibilità di sopravvivenza diminuiscono.
E molti di noi hanno le nostre teorie sul motivo per cui ciò accade. Ed è perché se siamo depressi, non diciamo: "Oh, mangerò dei cavoletti di Bruxelles, prenderò quelle vitamine del gruppo B, e uscirò a fare esercizio, e farò un po' di meditazione". Quindi il fattore di rischio indipendente post-IM per un evento è la depressione. La nostra mentalità riguardo alla depressione ci rende incapaci di funzionare normalmente e può far sviluppare al nostro corpo problemi che colpiscono i nostri organi vitali, muscoli e articolazioni. Quindi, la depressione gioca un ruolo importante, poiché il 75% dei decessi post-IM è correlato alla depressione, giusto? Quindi guardando i pazienti, ora, dovete porre la domanda: è la depressione che causa il problema, o è la malattia da citochine che ha già portato alla malattia cardiaca che causa la depressione? Dobbiamo tenere conto di tutto questo.
E ancora un altro studio ha esaminato oltre 4,000 persone senza malattia coronarica al basale. Per ogni aumento di cinque punti sulla scala della depressione, il rischio aumentava del 15%. E quelli con i punteggi di depressione più alti avevano un tasso di malattia coronarica più alto del 40% e un tasso di mortalità più alto del 60%. Quindi quasi tutti pensano che sia una malattia da citochine che porta a IM, malattie vascolari e depressione. E poi, ovviamente, quando hai un evento e ne esci fuori con tutta una serie di problemi intorno, sappiamo che le persone depresse hanno un doppio aumento della mortalità, un aumento di cinque volte della morte dopo un attacco di cuore, e scarsi risultati con la chirurgia. È così, cosa è venuto prima, la gallina o l'uovo?
In che modo la depressione è collegata allo stress cronico?
Il dottor Alex Jimenez, DC, presenta: Ogni chirurgo lo sa. Non vogliono fare interventi chirurgici su persone depresse. Sanno che il risultato non è buono e, naturalmente, è meno probabile che seguano tutte le nostre ottime raccomandazioni di medicina funzionale. Quindi quali sono alcuni dei meccanismi della disfunzione autonomica sono stati valutati variabilità della frequenza cardiaca e bassi livelli di omega-3, che hanno un profondo effetto sul cervello, e bassi livelli di vitamina D. Ci sono quelle citochine infiammatorie di cui abbiamo parlato che non ottengono sonno ristoratore e molti dei nostri cardiopatici soffrono di apnea. E ricorda, non pensare solo che siano i cardiopatici robusti con il collo corto e spesso; può essere abbastanza ingannevole. Ed è davvero importante osservare la struttura del viso e, ovviamente, la connessione sociale, che è la salsa segreta. Quindi la disfunzione autonomica è un meccanismo? Uno studio ha esaminato la variabilità della frequenza cardiaca nelle persone con un IM recente e hanno esaminato oltre 300 persone con depressione e senza depressione. Hanno scoperto che quattro indici di variabilità della frequenza cardiaca si abbasseranno nelle persone con depressione.
Infiammazione intestinale e stress cronico
Il dottor Alex Jimenez, DC, presenta: Quindi ecco due gruppi di persone che hanno un infarto e variabilità della frequenza cardiaca, che salgono in cima come possibile eziologia. Una delle tante cose che possono anche influenzare lo stress cronico nel corpo è il modo in cui il microbioma intestinale svolge la sua parte nello stress ossidativo. L'intestino è tutto e molti cardiopatici ridono perché chiedono ai loro cardiologi: "Perché ti interessa il mio microbioma intestinale? Perché questo dovrebbe influenzare il mio cuore? Bene, tutta quell'infiammazione intestinale sta causando la malattia da citochine. E quello che molti di noi hanno dimenticato dai tempi della scuola di medicina è che molti dei nostri neurotrasmettitori provengono dall'intestino. Quindi l'infiammazione cronica e l'esposizione alle citochine infiammatorie sembrano portare ad alterazioni della funzione della dopamina e dei gangli della base, riflesse da depressione, affaticamento e rallentamento psicomotorio. Quindi non possiamo enfatizzare abbastanza il ruolo dell'infiammazione e della depressione se diamo uno sguardo alla sindrome coronarica acuta e alla depressione, che era associata a marcatori più alti per l'infiammazione, CRP più elevata, HS inferiore, variabilità della frequenza cardiaca inferiore e qualcosa che mai viene controllato in ospedale, che è carenze nutrizionali.
E in questo caso, hanno esaminato i livelli di omega-3 e vitamina D, quindi come minimo, un controllo di omega-3 e un livello di vitamina D sono giustificati in tutti i nostri pazienti. E certamente, se riesci a ottenere una diagnosi completa per l'infiammazione indotta dallo stress. Un'altra condizione che devi considerare quando si tratta di infiammazione indotta dallo stress è l'osteoporosi delle articolazioni. Molte persone con osteoporosi avranno perdita muscolare, disfunzione immunitaria, grasso intorno alla linea mediana e glicemia alta sono associati all'invecchiamento e possono derivare da livelli elevati di cortisolo nel corpo.
I rischi elevati di malattie cardiache da cortisolo sono due volte superiori nelle persone che assumono alte dosi di steroidi. Piccole quantità di steroidi non hanno lo stesso rischio, quindi non è un grosso problema. Certo, cerchiamo di liberare i nostri pazienti dagli steroidi. Ma il punto qui è che il cortisolo è un ormone dello stress ed è un ormone dello stress che aumenta la pressione sanguigna e aumenta il peso sulla linea mediana, ci rende diabetici, provoca insulino-resistenza e l'elenco è infinito. Quindi, il cortisolo gioca un ruolo importante, e quando si tratta di medicina funzionale, dobbiamo esaminare i vari test che riguardano livelli elevati di cortisolo come la sensibilità alimentare, una valvola delle feci di 3 giorni, una nutra-valvola e uno stress surrenale indice di prova per vedere cosa sta succedendo con i pazienti. Quando c'è un sistema nervoso simpatico intensificato e cortisolo alto, abbiamo discusso di tutto, dalla coagulopatia alla diminuzione della variabilità della frequenza cardiaca, all'obesità centrale, al diabete e all'ipertensione.
Relazioni parentali e stress cronico
Il dottor Alex Jimenez, DC, presenta: E l'attivazione del sistema renina-angiotensina è tutto legato allo stress. Diamo un'occhiata a questo studio che ha esaminato 126 studenti di medicina di Harvard, e sono stati seguiti per 35 anni, una lunga ricerca. E hanno detto, qual è l'incidenza di malattie significative, malattie cardiache, cancro, ipertensione? E hanno posto a questi studenti domande molto semplici, qual era il tuo rapporto con tua madre e tuo padre? Era molto vicino? Era caloroso e amichevole? Era tollerante? Era teso e freddo? Questo è quello che hanno trovato. Hanno scoperto che se gli studenti identificavano la loro relazione con i loro genitori come un'incidenza tesa del 100% di un rischio per la salute significativo. Trentacinque anni dopo, se dicevano che era caldo e vicino, i risultati dimezzavano quella percentuale. E sarebbe d'aiuto se pensassi a cos'è e cosa può spiegarlo, e vedrai come le esperienze infantili avverse ci fanno ammalare in pochi minuti e come apprendiamo le nostre capacità di coping dai nostri genitori.
Conclusione
Il dottor Alex Jimenez, DC, presenta: La nostra tradizione spirituale viene spesso dai nostri genitori. I nostri genitori sono quelli che spesso ci insegnano come arrabbiarsi o come risolvere i conflitti. Quindi i nostri genitori hanno avuto un profondo effetto su di noi. E se ci pensi, anche la nostra connessione non è molto sorprendente. Questo è uno studio di follow-up di 35 anni.
Lo stress cronico può portare a molteplici problemi che possono essere correlati a malattie e disfunzioni nei muscoli e nelle articolazioni. Può colpire il sistema intestinale e portare a infiammazioni se non viene curato immediatamente. Quindi, quando si tratta dell'impatto dello stress sulla nostra vita quotidiana, possono essere numerosi fattori, dalle condizioni croniche alla storia familiare. Mangiare cibi nutrienti ricchi di antiossidanti, fare esercizio fisico, praticare la consapevolezza e sottoporsi a trattamenti quotidiani può ridurre gli effetti dello stress cronico e ridurre i sintomi associati che si sovrappongono e causano dolore al corpo. Possiamo continuare il nostro percorso di salute e benessere senza dolore utilizzando vari modi per ridurre lo stress cronico nel nostro corpo.
Il Dr. Alex Jimenez, DC, presenta come lo stress può avere un impatto su molti individui e correlarsi con molte condizioni nel corpo in questa serie in 2 parti. Indirizziamo i nostri pazienti a fornitori di servizi medici certificati che forniscono molteplici trattamenti disponibili per molte persone che soffrono di ipertensione associata al sistema cardiovascolare, endocrino e immunitario che colpisce il corpo. Incoraggiamo ciascuno dei nostri pazienti menzionandoli ai fornitori di servizi medici associati in base alla loro analisi in modo appropriato. Comprendiamo che l'educazione è un modo delizioso per porre domande ai nostri fornitori su richiesta e comprensione del paziente. Il dottor Jimenez, DC, utilizza queste informazioni solo come servizio educativo. Negazione di responsabilità
Come lo stress influisce sul corpo
Il dottor Alex Jimenez, DC, presenta: Ora ognuno risponde ai cambiamenti nell'ambiente in modo diverso. Quando si tratta di molte persone che svolgono attività quotidiane dal lavoro al lavoro, apertura nei fine settimana, ingorghi, sostenere esami o prepararsi per un discorso importante, il corpo passa attraverso uno stato costante di iperreattività a uno stadio di esaurimento emotivo e mentale che lascia l'individuo esausto e stressato. E la chiave è riconoscerlo prima che accada, poiché vediamo questo impatto dello stress sui nostri pazienti e su noi stessi. E la prima cosa da capire è quale sia l'evento scatenante che sta causando questo impatto.
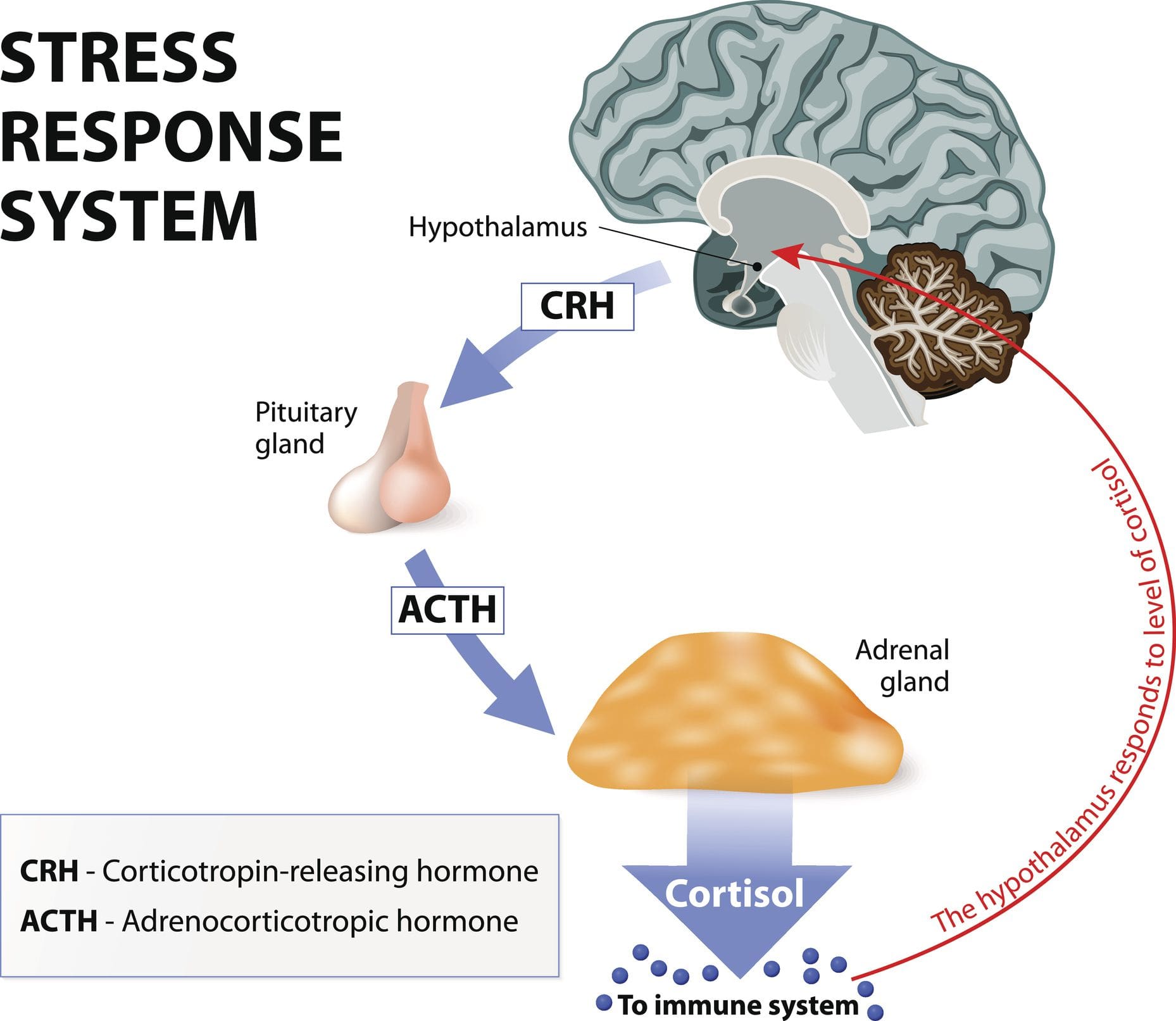
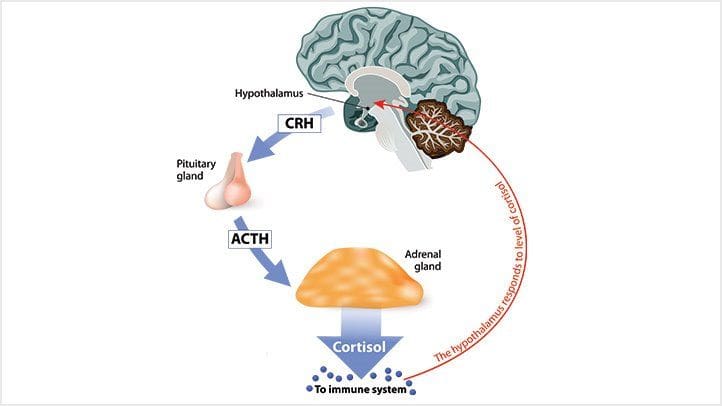
Qualunque sia l'evento scatenante, la parte più importante è la nostra percezione dell'evento. Cosa significa per noi? È la nostra percezione? Quando il corpo attraversa questo evento iniziale, può far sì che la percezione porti alla risposta e all'effetto sul nostro corpo. Quindi la percezione è tutto mentre parliamo di stress e della risposta allo stress. Ora, abbiamo oltre 1400 reazioni chimiche che si verificano nel corpo. Quindi, ai fini di questo discorso, discuteremo i tre fattori chiave: adrenalina e neuro-adrenalina, aldosterone e, naturalmente, cortisolo.
E perché sono importanti? Perché ognuno di questi ha un enorme impatto sulle malattie cardiovascolari. Ora, negli anni '1990, molti medici stavano iniziando a comprendere l'effetto dello stress sul corpo fisico. E cosa succede alle persone quando il loro asse HPA segnala che sono minacciate e iniziano a inondare i loro corpi di ormoni dello stress? Bene, vediamo una coagulazione migliorata. Vediamo uno spostamento nel sistema renina e angiotensina. Aumenta di giri. Vediamo aumento di peso nelle persone e insulino-resistenza. Quello che molte persone non capiscono è che i lipidi diventano anormali con lo stress. Quasi tutti i nostri pazienti sanno che la tachicardia e l'aritmia si verificano quando la nostra adrenalina scorre e la nostra pressione sanguigna aumenta. Ora, pensa a questo attraverso il linguaggio della medicina.
Intorno agli anni '1990, all'epoca i medici somministravano aspirina e Plavix per la coagulazione. Continuiamo a fornire ACE e ARB ai nostri pazienti. L'impatto del cortisolo provoca aumento di peso e resistenza all'insulina. Diamo statine; diamo metformina. Forniamo beta-bloccanti per quello, tachicardia e calcio-antagonisti per quella pressione alta. Quindi ogni singolo ormone che viene attivato con lo stress, abbiamo un farmaco che stiamo usando per bilanciarlo. E francamente, per anni, abbiamo parlato di quanto fossero buoni i beta-bloccanti per il cuore. Bene, quando ci pensi, i beta-bloccanti bloccano l'adrenalina. Quindi, quando i medici guardano questo, iniziano a pensare: “Beh, forse abbiamo bisogno di medicare e meditare, giusto? Usiamo tutti questi farmaci, ma potremmo aver bisogno di cercare altri modi per trasformare la risposta allo stress".
Cos'è la vasocostrizione?
Il dottor Alex Jimenez, DC, presenta: Non leggeremo ognuno di questi sintomi perché ce ne sono così tanti, ma tutto si riduce alla stessa cosa. Fatica. Dobbiamo pensare a qualcuno che ha avuto un incidente d'auto, per esempio, e quella persona sta sanguinando. Quindi il corpo è bello in quanto mette insieme un modo per fermare l'individuo dal sanguinamento o dalla vasocostrizione. La vasocostrizione sta costruendo questi vasi sanguigni e rendendo le piastrine appiccicose in modo che formino un coagulo e il sangue possa fermarsi. Ciò aumenta la gittata cardiaca aumentando la frequenza cardiaca e aumenta l'aldosterone, che provoca ritenzione di acqua e sale per aumentare la pressione sanguigna. Quindi per qualcuno in un'emergenza medica, come un incidente, sanguinamento o perdita di volume, questa è la bellezza del corpo umano. Ma sfortunatamente, vediamo persone che vivono in questo modo, letteralmente 24 ore su 7, XNUMX giorni su XNUMX. Quindi conosciamo la vasocostrizione e la viscosità piastrinica e vediamo aumenti dei marcatori di infiammazione, omocisteina, CRP e fibrinogeno, che aumentano il rischio cardiovascolare.
Vediamo l'impatto del cortisolo, non solo aumentando la pressione sanguigna, non solo causando diabete e insulino-resistenza, ma anche depositando grasso addominale intorno alla linea mediana. E poi, come vedrai tra pochi minuti, ci sono collegamenti tra eventi stressanti e aritmie come la fibrillazione atriale e persino la fibrillazione ventricolare. Per la prima volta in medicina, in cardiologia, abbiamo una sindrome chiamata cardiomiopatia di takosubo, affettuosamente chiamata sindrome del cuore spezzato. E questa è una sindrome in cui il miocardio diventa acutamente stordito al punto da causare una grave funzione o disfunzione del ventricolo sinistro. E di solito, questo è innescato da cattive notizie e da un evento emotivamente stressante. Sembra che qualcuno abbia bisogno di un trapianto di cuore. Quindi, quando pensiamo ai vecchi fattori di rischio di Framingham, diciamo, quali di questi sono influenzati dallo stress?
Sintomi di stress
Il dottor Alex Jimenez, DC, presenta: Le persone hanno tutti i tipi di comportamenti disadattivi da sottolineare, che si tratti di 20 amici in questo pacchetto di sigarette, mangiando questo Cinnabon perché mi fa sentire bene in questo momento, o tutto il cortisolo mi farà ingrassare e diventare diabetico. I lipidi salgono sotto stress; la pressione sanguigna sale sotto stress. Quindi ognuno di questi fattori di rischio è influenzato dagli ormoni dello stress. E, naturalmente, sappiamo che con l'attivazione del sistema RAS o del sistema renina-angiotensina, assistiamo sempre a un peggioramento dell'insufficienza cardiaca. E questo è molto descritto in letteratura. E, per quelli di voi che potrebbero lavorare al pronto soccorso, chiedete ai vostri pazienti cosa stavano facendo prima di entrare con il loro episodio di insufficienza cardiaca congestizia o dolore toracico. E ascolterai storie come, stavo guardando un brutto film, o stavo guardando un film di guerra, o mi sono arrabbiato per la partita di football, o qualcosa del genere.
Parleremo della variabilità della frequenza cardiaca, che viene influenzata dallo stress. E, naturalmente, lo stress influisce sulla nostra capacità di resistere alle infezioni. E sappiamo che le persone sono sotto stress quando vengono vaccinate. Ad esempio, i laser Cleco funzionano ma non producono anticorpi contro il vaccino quando sono sotto stress. E, naturalmente, come vedrete tra un minuto, un forte stress può causare morte cardiaca improvvisa, infarto del miocardio e così via. Quindi è un cattivo giocatore che viene trascurato. E per molti dei nostri pazienti, lo stress guida il treno. Quindi, quando parliamo di mangiare cavoletti di Bruxelles e cavolfiore e, sai, un sacco di verdure a foglia verde, e qualcuno è così stressato che sta cercando di capire: “Come farò a superare la giornata? " Non sentono nessuna delle altre cose che raccomandiamo.
Quindi, lo stress cronico e i disturbi affettivi, che siano depressione, ansia o panico, mettono il piede sull'acceleratore e mandano su di giri il sistema nervoso simpatico. Sappiamo che le stesse cose che vediamo con l'invecchiamento, come vedrai tra un minuto, sono legate all'aumento dei livelli di ormoni dello stress, in particolare il cortisolo. Quindi, che si tratti di osteoporosi, diminuzione della densità ossea, disfunzione endoteliale, attivazione piastrinica, ipertensione, obesità centrale o insulino-resistenza, questo deriva da una risposta allo stress. E dobbiamo avere un piano per i nostri pazienti su come gestirlo. L'American Institute of Stress afferma che dal 75 al 90% di tutte le visite degli operatori sanitari derivano da disturbi legati allo stress. Ed è troppo alto, ma guardando i pazienti e dove stavano arrivando, raccontano le loro storie ai loro dottori. I risultati sono gli stessi; non importa se si trattava di mal di testa, tensione muscolare, angina, aritmia o intestino irritabile; ha quasi sempre avuto qualche fattore scatenante dello stress.
Stress acuto e cronico
Il dottor Alex Jimenez, DC, presenta: C'è una differenza tra stress acuto e cronico con la nostra percezione e connessione sociale. Anche se otteniamo un po' di forza da un potere superiore, lo stress può avere un impatto su chiunque e la maggior parte di noi potrebbe non essere in grado di gestirlo bene. Quindi un grande studio è stato fatto molti anni fa dal Dr. Ray e Holmes che affermava, 50 anni fa, messo insieme un metodo per quantificare gli eventi che cambiano la vita. Quindi diamo un'occhiata ad alcune aree, come gli eventi che cambiano la vita. In che modo gli eventi che cambiano la vita e come si classificano? Quali sono i grandi e quali i piccoli?
E in che modo questa classifica porta a gravi problemi medici come cancro, infarto e morte improvvisa in futuro? Quindi hanno esaminato 43 eventi che cambiano la vita, li hanno classificati in origine e li hanno riclassificati negli anni '1990. E alcuni di loro sono rimasti gli stessi. Hanno assegnato un punteggio di aggiustamento all'evento e poi hanno esaminato i numeri che sarebbero stati collegati a malattie gravi. Quindi, ad esempio, un evento che cambia la vita. Il numero uno, 100 unità che cambiano la vita, è la morte di un coniuge. Chiunque potrebbe identificarsi con quello. Il divorzio era il numero due, la separazione il numero tre e la fine di un familiare stretto. Ma ho anche notato che alcune cose sono state classificate che potrebbero non essere equiparate a essere un importante evento che cambia la vita che può avere un impatto su una risposta allo stress come il matrimonio o il pensionamento.
Conclusione
Il dottor Alex Jimenez, DC, presenta: Quindi non è stato il singolo evento a fare la differenza. Era la somma degli eventi. E quello che hanno scoperto dopo aver esaminato 67 medici è stato che se avevi un punteggio unitario che cambia la vita da qualche parte tra zero e uno 50, non è un grosso problema, nessuna vera malattia grave, ma una volta raggiunto quel limite di 300, c'era un 50% possibilità di malattia grave. Quindi questa cronologia degli eventi nella vita del paziente. Vogliamo sapere cosa stava succedendo nella loro vita quando sono iniziati i sintomi e poi riportarlo prima per capire l'ambiente in cui viveva questo individuo. L'impatto dello stress può far sviluppare a molte persone condizioni croniche e mascherare altri sintomi che possono portare a dolori muscolari e articolari. Nella parte 2, approfondiremo il modo in cui l'impatto dello stress influisce sul corpo e sulla salute di una persona.
Dr. Alex Jimenez, DC, presenta come l'ipertensione colpisce il corpo umano e alcune cause che possono aumentare l'ipertensione in molti individui in questa serie in 2 parti. Indirizziamo i nostri pazienti a fornitori di servizi medici certificati che forniscono molteplici trattamenti disponibili per molte persone che soffrono di ipertensione associata al sistema cardiovascolare e immunitario che colpisce il corpo. Incoraggiamo ciascuno dei nostri pazienti menzionandoli ai fornitori di servizi medici associati in base alla loro analisi in modo appropriato. Comprendiamo che l'educazione è un modo delizioso per porre domande ai nostri fornitori su richiesta e comprensione del paziente. Il Dr. Jimenez, DC, utilizza queste informazioni solo come servizio educativo. Negazione di responsabilità
Come cercare l'ipertensione
Il dottor Alex Jimenez, DC, presenta: Torniamo all'albero decisionale in modo che possiate iniziare a pensare a come applicherete il modello go-to-it nella medicina funzionale all'ipertensione e come inizierete a valutare meglio qualcuno con ipertensione piuttosto che dire loro che la loro pressione sanguigna è elevata . Il corpo è influenzato da infiammazione, stress ossidativo o risposta immunitaria? Sta influenzando la funzione endoteliale o la muscolatura liscia vascolare da queste tre categorie di reazioni, infiammazione, stress ossidativo o risposta immunitaria? Scegliamo un calcioantagonista diuretico o un ACE inibitore? E quindi per farlo, è davvero importante nella nostra sezione di raccolta. Prendendo la storia medica e la sequenza temporale della loro ipertensione, dai questionari ottieni un indizio sul danno d'organo. Stai guardando i loro dati antropometrici.
Ciò include le seguenti domande:
Quali sono i marcatori infiammatori?
Quali sono i biomarcatori e gli indicatori clinici?
Questi sono delineati attraverso l'albero decisionale clinico. E già solo facendo questo, espanderai e perfezionerai la tua lente su ciò che potresti vedere nel tuo paziente iperteso. Aggiungiamo alla cronologia quando inizia l'ipertensione? Il lasso di tempo dell'ipertensione inizia effettivamente nel periodo prenatale. È importante chiedere al paziente se era in età precoce o in età scolare. La loro madre era stressata? Sono nati prematuri o prematuri? C'è stato stress nutrizionale durante la gravidanza? Se lo sanno, puoi avere due persone con la stessa dimensione renale, ma la persona che non ha avuto abbastanza proteine durante la gravidanza può avere fino al 40% in meno di glomeruli. Sapere che cambierà il modo in cui aggiusti il farmaco decenni dopo se sai che forse hanno il 40% in meno di glomeruli.
La cronologia per la pressione sanguigna
Il dottor Alex Jimenez, DC, presenta: Quindi è importante prendere la cronologia della loro pressione sanguigna. Poi è anche importante riconoscere cosa sta succedendo quando iniziamo a organizzare e raccogliere dati attraverso i biomarcatori; i biomarcatori di base ti daranno indizi sul fatto che abbiano problemi con i lipidi dell'insulina, se hanno problemi con la reattività vascolare, l'equilibrio del sistema nervoso autonomo, lo squilibrio, la coagulazione o gli effetti delle tossine immunitarie. Quindi questa è una cosa ragionevole da stampare perché, nel tuo paziente iperteso, è solo attraverso i biomarcatori che puoi iniziare a ottenere un indizio su quali aree di disfunzione influenzano l'infiammazione, lo stress ossidativo e la risposta immunitaria e come questi biomarcatori lo riflettono informazioni per te. Questo è molto ragionevole da avere davanti a te per aiutarti a cambiare i tuoi pensieri sull'ipertensione e ti consente anche di affinare alcune delle caratteristiche della persona dall'altra parte del tuo stetoscopio in un modo più personalizzato e preciso.
Ma cominciamo dall'inizio. Il tuo paziente ha la pressione alta? Sappiamo che a seconda degli effetti degli organi terminali delle loro comorbidità, potresti avere una pressione sanguigna leggermente più alta se hai un problema di profusione nel cervello, nei reni o nel cuore, ma ci sono alcune linee guida. Le nostre linee guida 2017 dell'American Heart Association per le categorie di pressione sanguigna sono elencate qui. Sono aumentati e diminuiti avanti e indietro negli ultimi due decenni, ma questo è molto chiaro. Avere una pressione sanguigna elevata, qualsiasi cosa al di sopra di 120, ha davvero cambiato il numero di persone che iniziamo a vedere o considerare di affrontare le cause alla radice della loro pressione sanguigna. Quindi torneremo su questo, specialmente nel caso per aiutarci a vedere come classifichiamo le persone con problemi di pressione sanguigna.
I criteri per misurare la pressione sanguigna
Il dottor Alex Jimenez, DC, presenta: Qual è il primo passo? È come fai a misurare la pressione sanguigna al tuo paziente? Lo controllano a casa? Ti portano quei numeri? Come monitorate la pressione sanguigna nella vostra clinica? Come si ottengono letture accurate nella propria clinica? Ecco i criteri per misurare con precisione la pressione sanguigna e le domande da considerare se stai facendo tutto questo.
Chiedi al tuo paziente se ha assunto caffeina nell'ultima ora?
Se hanno fumato nell'ora precedente?
Sono stati esposti al fumo nell'ultima ora?
Il luogo in cui stai misurando la pressione sanguigna è caldo e tranquillo?
Sono seduti con la schiena appoggiata su una sedia con i piedi per terra?
Usi il tavolino girevole per appoggiare il braccio all'altezza del cuore?
Sono seduti al tavolo degli esami con i piedi penzolanti e un assistente infermiere alza il braccio e mette la piega ascellare per tenergli il braccio lì?
Hanno i piedi per terra?
Sono rimasti seduti lì per cinque minuti?
Si sono esercitati nei 30 minuti precedenti?
Potresti avere la pressione arteriosa sistolica se tutto rientra nei criteri. Ecco la sfida. Ci sono da 10 a 15 millimetri di mercurio in più quando si sta seduti e si misura la pressione sanguigna. E la misura del polsino? Conosciamo il secolo scorso; la maggior parte degli adulti aveva una circonferenza della parte superiore del braccio inferiore a 33 centimetri. Oltre il 61% delle persone ora ha una circonferenza del braccio superiore a 33 centimetri. Quindi la dimensione del bracciale è diversa per circa il 60% dei pazienti adulti, a seconda della popolazione. Quindi devi usare un polsino grande. Quindi dai un'occhiata a come viene raccolta la pressione sanguigna nel tuo ufficio. Diciamo che la pressione sanguigna è elevata nei tuoi pazienti; allora dobbiamo chiedere, è normale? Grande.
I diversi tipi di ipertensione
Il dottor Alex Jimenez, DC, presenta: È elevato a causa dell'ipertensione da camice bianco? Hanno una pressione sanguigna normale, elevata al di fuori della clinica o ipertensione mascherata? O hanno solo un'ipertensione sostenuta che è una sfida? Ne parleremo. Quindi, quando interpreti, è anche importante considerare il monitoraggio ambulatoriale della pressione arteriosa. Quindi, se hai qualcuno che è iperteso e non sai se la pressione sanguigna scende e stai cercando di capire se ha ipertensione sostenuta, puoi usare il monitoraggio della pressione sanguigna 24 ore su 130. La pressione arteriosa media diurna superiore a 80 su 110 è ipertensiva, la pressione arteriosa notturna media superiore a 65 su 15 è ipertensiva. Quindi perchè è importante? La pressione sanguigna media scende a circa il XNUMX% durante la notte a causa del problema con l'abbassamento della pressione sanguigna. Il mancato calo della pressione sanguigna durante il sonno notturno potrebbe sviluppare problemi che possono interessare una persona durante il giorno.
Se il tuo paziente dorme di notte, dovrebbe diminuire di circa il 15% quando dorme. Se hanno una pressione sanguigna non discendente, è associata a comorbilità. Quali sono alcune di queste comorbilità nella pressione sanguigna non abbassata? Alcune delle condizioni correlate con la pressione sanguigna senza immersione includono:
Malattia cardiaca congestizia
Malattie Cardiovascolari
Malattia cerebrovascolare
Insufficienza cardiaca congestizia
Fallimento renale cronico
Infrazioni cerebrali silenziose
Co-morbidità associate a pressione non sanguigna
Il dottor Alex Jimenez, DC, presenta: Queste sono le comorbilità associate alla pressione non sanguigna. Siamo tutti d'accordo sul fatto che la pressione sanguigna elevata non è necessariamente buona in tutte quelle condizioni. Quindi, quando si osservano diversi gruppi di persone o altre comorbidità, la pressione arteriosa non abbassata è più comunemente associata a persone sensibili al sodio, persone che hanno insufficienza renale, persone che hanno il diabete, persone che hanno l'ipertrofia ventricolare sinistra, persone che hanno ipertensione refrattaria o disfunzione del sistema nervoso autonomo e, infine, apnea notturna. Quindi, la pressione sanguigna non abbassata aumenta la tua associazione con un danno cardiaco subclinico. Ok, l'immersione inversa significa che sei più iperteso di notte ed è più associato all'ascesa che durante il giorno è più correlato all'ictus emorragico. E se hai qualcuno con ipertensione notturna, devi iniziare a pensare a cose come le arterie carotidi e l'aumento dello spessore mediale interno della carotide. Inizi a pensare all'ipertrofia ventricolare sinistra e potresti vederlo sull'ECG. Ecco cosa sappiamo dell'ipertensione notturna. L'ipertensione notturna è una pressione arteriosa notturna superiore a 120 su 70. È associata a una maggiore prevedibilità di morbilità e mortalità cardiovascolare.
Se soffri di ipertensione notturna, aumenta il rischio di mortalità per malattie cardiovascolari dal 29 al 38%. Dobbiamo sapere cosa succede di notte quando dormiamo, giusto? Bene, qual è un'altra raffinatezza? Un altro perfezionamento è riconoscere che la pressione sanguigna a riposo è controllata dal sistema renina-angiotensina. La pressione sanguigna al risveglio è controllata dal sistema nervoso simpatico. Quindi parliamo di come il loro sistema renale di angiotensina guida la loro ipertensione notturna e pensi a quali farmaci stanno assumendo. Potresti cambiare il dosaggio del farmaco alla notte. Bene, gli studi hanno dimostrato che se hai l'ipertensione notturna e non sei un merlo acquaiolo, è meglio prendere i tuoi ACE-inibitori, ARB, calcio-antagonisti e alcuni beta-bloccanti la sera prima di andare a letto. Ma ha senso che tu non sposti i tuoi diuretici alla notte, o avrai un sonno disturbante.
Affrontare la pressione sanguigna diurna e notturna
Il dottor Alex Jimenez, DC, presenta: Quindi, se non affrontiamo la pressione sanguigna diurna e notturna, dobbiamo considerare l'effetto del carico pressorio. Qual è la tua pressione sanguigna media diurna e la tua pressione sanguigna moderata durante il sonno. Sappiamo che il carico pressorio nei giovani adulti è ipertensivo solo circa il 9% delle volte. Ciò significa che il carico sistolico è di circa il 9% rispetto agli anziani, circa l'80% del carico di pressione sanguigna è sistolico. E così quando hai un carico sistolico più alto, hai più complicazioni e danni agli organi terminali. Quindi quello di cui stiamo parlando è aiutare a identificare il tuo paziente con ipertensione; qual è la loro cronologia? Qual è il loro fenotipo? Sono ipertesi solo di giorno o anche di notte? Dobbiamo guardare a cosa aiuta a bilanciarlo.
Ecco l'altro punto, solo circa il 3.5% delle persone con ipertensione ha una causa genetica. Solo il 3.5% delle persone i loro geni causano l'ipertensione. Il potere è alla base della matrice e riconosce questi schemi, giusto? Quindi guardi l'esercizio, il sonno, la dieta, lo stress e le relazioni. Quindi sappiamo che questi quattro equilibri autonomici aiutano a determinare la pressione sanguigna. Esamineremo il sistema renale dell'angiotensina, il volume plasmatico in cui trattengono troppo fluido, il carico salino secondario e la disfunzione endoteliale. Anomalie in uno qualsiasi di questi possono portare all'ipertensione. Abbiamo parlato di un altro che può portare all'ipertensione: il legame tra insulino-resistenza e ipertensione.
Questo schematicamente ti dà un'idea delle interazioni fisiologiche tra insulino-resistenza e ipertensione. Colpisce l'aumento del tono simpatico e l'aumento dell'equilibrio del sistema renale-angiotensina. Quindi dedichiamo qualche minuto al percorso del sistema renina-angiotensina dall'angiotensinogeno fino all'angiotensina due. Approfittiamo di questi enzimi somministrando inibitori agli enzimi di conversione dell'angiotensina nei nostri pazienti ipertesi. L'angiotensina due elevata porta all'ipertrofia cardiovascolare, porta alla costrizione della fase simpatica, all'aumento del volume sanguigno, al fluido di sodio, alla ritenzione e al rilascio di aldosterone. Puoi chiedere informazioni sui biomarcatori dei tuoi pazienti? Puoi chiedere se hanno livelli elevati di renina?
Cerca i segni
Il dottor Alex Jimenez, DC, presenta: Beh, puoi. È possibile controllare l'attività della renina plasmatica e i livelli di aldosterone. È importante farlo se il paziente è iperteso e non ha mai assunto farmaci perché è qui che il protossido di azoto è così importante. È qui che è presente la tua ossido nitrico sintasi endoteliale. Qui è dove hai lo stress puro ed emodinamico. È qui che l'assunzione alimentare di arginina o l'ambiente che influenza l'ossido nitrico gioca un ruolo importante nella salute di questo strato di endotelio. Se metti tutto insieme in qualche modo, miracolosamente, o almeno con gli occhi della tua mente, coprirà sei campi da tennis nell'adulto medio. È una superficie enorme. E le cose che causano la disfunzione endoteliale non sono una novità per le persone in medicina funzionale. L'aumento dello stress ossidativo e dell'infiammazione sono due cose che abbiamo menzionato che hanno un effetto.
E poi, guarda alcuni di questi altri componenti, il tuo ADMA è elevato e correlato alla resistenza all'insulina. Tutto comincia a formarsi insieme in una matrice che interagisce. Quindi guardi una comorbidità nella sindrome cardiometabolica e colpisce un'altra comorbidità. All'improvviso vedi l'interrelazione tra loro o l'iperomocisteinemia, che è un marcatore del metabolismo a un carbonio, il che significa che stai osservando l'adeguatezza di folati, b12, b6, riboflavina e quell'attività del tuo metabolismo a un carbonio. Quindi diamo un'occhiata ad alcuni di questi marcatori di rischio emergenti per migliorare e monitorare i pazienti con ipertensione. Rianalizziamo nuovamente l'ADMA. ADMA sta per dimetil arginina asimmetrica. Asimmetrica, la dimetilarginina è un biomarcatore della disfunzione endoteliale. Quella molecola inibisce l'ossido nitrico sintasi compromettendo la funzione endoteliale e in tutte le comorbidità associate alla sindrome cardiometabolica, l'ADMA può essere elevata.
Conclusione
Quindi, come rapida rassegna, la L-arginina viene convertita in ossido nitrico tramite l'ossido nitrico sintasi e l'adeguatezza dell'ossido nitrico porta alla vasodilatazione. ADMA blocca questa conversione. E se i tuoi livelli di ADMA sono elevati e i tuoi livelli di ossido nitrico sono bassi, allora hai diminuito l'aumento dell'aggregazione piastrinica dell'ossido nitrico nell'ossidazione delle LDL. Così tante cose riducono l'ossido nitrico o sono associate a livelli più bassi di ossido nitrico, apnea notturna, arginina dietetica bassa, proteine, insufficienza di zinco e fumo.
Tutti si occupano stress ad un certo punto della loro vita. Che si tratti di un colloquio di lavoro, di una scadenza enorme, di un progetto o anche di un test, lo stress è lì per mantenere il corpo funzionante in ogni scenario che il corpo sta attraversando. Lo stress può aiutare a regolare il corpo sistema immunitario e aiuto metabolizzare l'omeostasi poiché il corpo aumenta la sua energia durante il giorno. Quando si ha a che fare con stress cronico può causare disfunzioni metaboliche nel corpo come disturbi intestinali, infiammazioni e un aumento dei livelli di glucosio nel sangue. Lo stress cronico può anche influenzare l'umore e la salute di una persona, le abitudini alimentari e la qualità del sonno. L'articolo di oggi esaminerà se lo stress è positivo o negativo, come influisce sul corpo e gli effetti di ciò che lo stress cronico fa al corpo. Indirizzare i pazienti a fornitori certificati e qualificati specializzati in trattamenti intestinali per individui che soffrono di neuropatia autonomica. Guidiamo i nostri pazienti facendo riferimento ai nostri fornitori di servizi medici associati in base al loro esame quando è appropriato. Riteniamo che l'istruzione sia fondamentale per porre domande approfondite ai nostri fornitori. Il Dr. Alex Jimenez DC fornisce queste informazioni solo come servizio educativo. Negazione di responsabilità
La mia assicurazione può coprirlo? Sì, può. Se sei incerto, ecco il link a tutti i fornitori di assicurazioni che copriamo. In caso di domande o dubbi, chiamare il Dr. Jimenez al 915-850-0900
Avere lo stress fa bene o male?
Ti senti sempre ansioso? Che ne dici di provare mal di testa che sono costantemente una seccatura? Ti senti sopraffatto e perdi concentrazione o motivazione? Tutti questi segni sono situazioni stressanti che una persona sta attraversando. Gli studi di ricerca hanno definito stress o cortisolo come ormone del corpo che fornisce una varietà di effetti su diverse funzioni in ciascun sistema. Il cortisolo è il glucocorticoide primario che proviene dalla corteccia surrenale. Allo stesso tempo, l'asse HPA (ipotalamo-ipofisi-surrene) aiuta a regolare la produzione e la secrezione di questo ormone al resto del corpo. Ora il cortisolo può essere benefico e dannoso per il corpo, a seconda della situazione in cui si trova una persona. Ulteriori studi di ricerca hanno menzionato che il cortisolo inizia e colpisce il cervello e il resto del corpo poiché lo stress nella sua forma acuta può indurre il corpo ad adattarsi e sopravvivere. Le risposte acute del cortisolo consentono la funzione neurale, cardiovascolare, immunitaria e metabolica nel corpo.
In che modo influisce sul metabolismo del corpo?
Ora il cortisolo influenza il metabolismo del corpo quando viene controllato in un ciclo del sonno lento e costante che diminuisce l'ormone di rilascio della corticotropina (CRH) e aumenta l'ormone della crescita (GH). Studi di ricerca hanno dimostrato che quando le ghiandole surrenali secernono cortisolo, inizia ad avere una complessa interazione con l'ipotalamo e le ghiandole pituitarie nel sistema nervoso ed endocrino. Ciò fa sì che la funzione surrenale e tiroidea nel corpo sia strettamente collegata mentre è sotto il controllo dell'ipotalamo e degli ormoni tropici. La tiroide compete con gli organi surrenali per la tirosina. Studi di ricerca hanno trovato che la tirosina viene utilizzata per produrre cortisolo sotto stress prevenendo il declino della funzione cognitiva che risponde allo stress fisico. Tuttavia, quando il corpo non è in grado di produrre abbastanza tirosina, può causare ipotiroidismo e causare la cronicizzazione dell'ormone cortisolo.
Una panoramica sullo stress-Video
Hai avuto mal di testa che si manifestano casualmente dal nulla? Hai costantemente guadagnato peso o perso peso? Ti senti sempre ansioso o stressato perché influisce sul tuo sonno? Questi sono tutti segni e sintomi dei livelli di cortisolo che si stanno trasformando nel loro stato cronico. Il video sopra mostra cosa fa lo stress al tuo corpo e come può causare sintomi indesiderati. Quando c'è stress cronico nel corpo, l'asse HPA (neuro-endocrino) è sbilanciato a causa degli attivatori mediati dallo stress coinvolti nelle malattie autoimmuni della tiroide (AITD). Quando c'è stress cronico nel corpo, può causare un'eccessiva produzione di composti infiammatori nel corpo che possono generare IR. Le sostanze infiammatorie possono danneggiare o inattivare i recettori dell'insulina portando all'insulino-resistenza. Ciò contribuisce quindi alla rottura di uno o più fattori necessari per completare il processo di trasporto del glucosio nel corpo.
Gli effetti del cortisolo cronico nel corpo
Quando c'è stress cronico nel corpo e non è stato trattato o ridotto subito, può portare a qualcosa noto come carico allostatico. Il carico allostatico è definito come l'usura del corpo e del cervello dovuta all'iperattività cronica o all'inattività dei sistemi corporei tipicamente coinvolti nelle sfide ambientali e nell'adattamento. Studi di ricerca hanno dimostrato quel carico allostatico provoca un'eccessiva secrezione di ormoni come il cortisolo e la catecolamina per rispondere a fattori di stress cronici che colpiscono il corpo. Ciò fa sì che l'asse HPA faccia una delle due cose: essere sovraccarico di lavoro o non riuscire a spegnersi dopo eventi stressanti che causano disturbi del sonno. Altri problemi che lo stress cronico causa al corpo possono includere:
Aumento della secrezione di insulina e deposito di grasso
Funzione immunitaria alterata
Ipotiroidismo (esaurimento surrenale)
Ritenzione di sodio e acqua
Perdita del sonno REM
Instabilità mentale ed emotiva
Aumento dei fattori di rischio cardiovascolare
Questi sintomi fanno sì che il corpo diventi disfunzionale e hanno evidenziato studi di ricerca che vari fattori di stress possono danneggiare il corpo. Questo può rendere estremamente difficile per una persona far fronte allo stress e alleviarlo.
Conclusione
Nel complesso, lo stress o il cortisolo è un ormone di cui il corpo ha bisogno per funzionare correttamente. Lo stress cronico nel corpo dovuto a vari fattori di stress può causare molte disfunzioni metaboliche come ipotiroidismo, aumento di peso, insulino-resistenza e sindrome metabolica, solo per citarne alcuni. Lo stress cronico può anche causare disturbi del sonno poiché l'asse HPA è cablato e può sembrare minimamente calmarsi. Quando le persone iniziano a trovare modi per affrontare questi vari fattori di stress, possono ridurre i loro livelli di stress alla normalità ed essere senza stress.
Riferimenti
Jones, Carol e Christopher Gwenin. "Disregolazione del livello di cortisolo e sua prevalenza: è la sveglia della natura?" Relazioni fisiologiche, John Wiley and Sons Inc., gennaio 2021, www.ncbi.nlm.nih.gov/pmc/articles/PMC7749606/.
McEwen, Bruce S. "Effetti centrali degli ormoni dello stress nella salute e nella malattia: comprensione degli effetti protettivi e dannosi dello stress e dei mediatori dello stress". European Journal of Pharmacology, Biblioteca nazionale di medicina degli Stati Uniti, 7 aprile 2008, www.ncbi.nlm.nih.gov/pmc/articles/PMC2474765/.
McEwen, Bruce S. "Stressato o stressato: qual è la differenza?" Journal of Psychiatry & Neuroscience: JPN, Biblioteca nazionale di medicina degli Stati Uniti, settembre 2005, www.ncbi.nlm.nih.gov/pmc/articles/PMC1197275/.
Rodriquez, Erik J, et al. "Carico allostatico: importanza, indicatori e determinazione del punteggio nelle popolazioni di minoranze e disparità". Journal of Urban Health: Bollettino della New York Academy of Medicine, Springer USA, marzo 2019, www.ncbi.nlm.nih.gov/pmc/articles/PMC6430278/.
Thau, Lauren, et al. "Fisiologia, cortisolo - Statpearls - Libreria NCBI". In: StatPearls [Internet]. L'isola del tesoro (FL), StatPearls Publishing, 6 settembre 2021, www.ncbi.nlm.nih.gov/books/NBK538239/.
Young, Simon N. "L-tirosina per alleviare gli effetti dello stress?" Journal of Psychiatry & Neuroscience: JPN, Biblioteca nazionale di medicina degli Stati Uniti, maggio 2007, www.ncbi.nlm.nih.gov/pmc/articles/PMC1863555/.
Poiché il mondo è in continuo movimento, molte persone devono sopportare situazioni stressanti che intaccano il loro corpo e la loro salute. Il corpo ha bisogno di ormoni come cortisolo per continuare a funzionare in quanto influisce sul sistema immunitario, nervoso, cardiovascolare e muscolo-scheletrico, per dirne alcuni. Un'altra funzione essenziale di cui il corpo ha bisogno è il glucosio, che richiede energia per essere in costante movimento. Le situazioni che causano un aumento dei livelli di cortisolo e di glucosio nel corpo possono portare a problemi cronici come il diabete e lo stress cronico. Ciò fa sì che l'individuo sia infelice e si trovi in una situazione seria se non viene controllato immediatamente. L'articolo di oggi esamina come il cortisolo e il glucosio influenzano il corpo e la connessione intrecciata tra stress e diabete. Indirizzare i pazienti a fornitori qualificati e certificati, specializzati nella gestione dello stress e nei trattamenti endocrini per i soggetti diabetici. Guidiamo i nostri pazienti facendo riferimento ai nostri fornitori di servizi medici associati in base al loro esame quando è appropriato. Riteniamo che l'istruzione sia fondamentale per porre domande approfondite ai nostri fornitori. Il Dr. Alex Jimenez DC fornisce queste informazioni solo come servizio educativo. Negazione di responsabilità
La mia assicurazione può coprirlo? Sì, può. Se sei incerto, ecco il link a tutti i fornitori di assicurazioni che copriamo. In caso di domande o dubbi, chiamare il Dr. Jimenez al numero 915-850-0900.
In che modo il cortisolo influisce sul corpo?
Hai avuto problemi di sonno durante la notte? Che dire dei frequenti mal di testa che sono una seccatura per l'intera giornata? O hai notato un'eccessiva perdita di peso o aumento di peso intorno al tuo tronco? Alcuni di questi sintomi sono segni che i livelli di cortisolo e glucosio sono alti e possono influenzare il tuo corpo. Il cortisolo è un ormone prodotto nel sistema endocrino e può essere benefico o dannoso per l'organismo se non viene controllato regolarmente. Studi di ricerca hanno definito il cortisolo come uno dei principali glucocorticoidi secreti a causa della risposta delle sostanze biochimiche del corpo, caratterizzato dall'asse HPA (ipotalamo-ipofisi-surrene) aiuta gli eventi cognitivi. Tuttavia, quando i livelli di cortisolo diventano cronici nel corpo a causa di circostanze che fanno sì che il corpo diventi disfunzionale, può avere un impatto significativo su una persona e causare uno squilibrio nell'asse HPA. Alcuni dei sintomi che il cortisolo cronico porta al corpo possono includere:
Squilibri ormonali
Resistenza all'insulina
Aumento di peso
Aumento del grasso viscerale della "pancia".
Aumento della produzione di cortisolo
Problemi immunitari
Allergie e asma
Articolazioni infiammate
Scarso recupero dell'esercizio
Ulteriori informazioni sono state fornite che la presenza di cortisolo nel corpo può aiutare ad aumentare la disponibilità di glucosio nel sangue al cervello. Con il cortisolo che fornisce funzionalità d'organo, il glucosio nel sangue fornisce energia per il corpo.
Come funzionano il cortisolo e il glucosio nel corpo
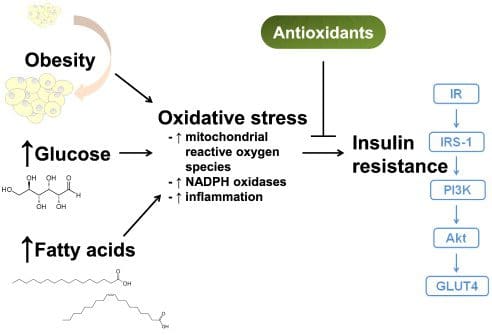
Il cortisolo aiuta a stimolare la mobilizzazione di massa del glucosio nel fegato, consentendo alla sintesi proteica di bloccare di spingere gli aminoacidi nello zucchero per il corpo. Questo è noto come liberazione di acidi grassi biotrasformata in glucosio. Quando ciò accade, aiuta a stimolare l'accumulo di grasso viscerale se non viene utilizzato il glucosio in eccesso, causando così un aumento di peso. Studi di ricerca hanno dimostrato che una mancanza di cortisolo può causare una diminuzione della produzione di glucosio epatico nel corpo. Ciò causerà ipoglicemia, in cui il corpo non ha abbastanza glucosio nel suo sistema. Ulteriori ricerche mostrano che il cortisolo risponda a qualsiasi fattore di stress che colpisce una persona con bassi livelli di glucosio ma può anche diventare positivo dopo un carico di glucosio. Gestire i livelli di glucosio e cortisolo nel corpo può aiutare a far progredire lo sviluppo del diabete.
Come il cortisolo è collegato al diabete di tipo 2 - Video
Hai vissuto situazioni stressanti che causano tensioni muscolari? Che ne dici di sentire il livello di zucchero nel sangue aumentare o diminuire? Senti effetti infiammatori su tutto il corpo che li fanno soffrire? Lo stress può causare effetti dannosi al corpo, attivando l'infiammazione, aumentando il tono simpatico e riducendo la reattività ai glucocorticoidi. Lo stress può anche essere collegato al diabete, poiché il video sopra mostra come l'ormone dello stress cortisolo è collegato al diabete di tipo 2. Gli studi di ricerca hanno menzionato che il cortisolo può associarsi negativamente alla meccanica dell'insulino-resistenza, aumentando la funzione delle cellule beta e aumentando l'insulina rilasciata nell'organismo. Questo può diventare pericoloso per molte persone che hanno il diabete preesistente e hanno affrontato costantemente lo stress.
La connessione intrecciata tra stress e diabete
Viene mostrata la connessione intrecciata tra stress e diabete hanno scoperto studi di ricerca che la fisiopatologia dell'ansia e del diabete ha aumentato il rischio di insulino-resistenza per l'organismo. Quando una persona ha a che fare con lo stress cronico, può causare molti problemi come:
Intolleranza fredda
Cognizione e umore diminuiti
Sensibilità alimentari
Bassa energia per tutto il giorno
Quando ciò accade, il corpo è ad alto rischio di sviluppare insulino-resistenza e diabete di tipo 2. Gli studi di ricerca hanno menzionato che il diabete di tipo 2 è caratterizzato da insulino-resistenza e disfunzione delle cellule beta. Il glucocorticoide nel corpo può diventare eccessivo per influenzare le cellule, causando disfunzioni. Ulteriori studi di ricerca hanno dimostrato che qualsiasi stress percepito può diventare un fattore di rischio vitale che non solo colpisce il corpo, come l'ipertensione, l'indice di massa corporea (indice di massa corporea) o la qualità della dieta, ma può causare un aumento del diabete di tipo 2. Quando le persone trovano il modo di ridurre il loro stress cronico, possono aiutare a gestire i loro livelli di glucosio dal raggiungere livelli critici.
Conclusione
Lo stress cronico del corpo può causare insulino-resistenza e far sì che il diabete diventi preesistente. Il corpo ha bisogno di cortisolo e glucosio per continuare a funzionare e avere l'energia per muoversi. Quando le persone iniziano a soffrire di stress cronico e diabete, può diventare difficile da gestire; tuttavia, apportare piccole modifiche al corpo come trovare modi per ridurre lo stress, mangiare cibi sani e monitorare i livelli di glucosio può aiutare il corpo a riportare i livelli di glucosio e cortisolo alla normalità. Fare questo può alleviare molte persone che vogliono continuare il loro viaggio verso la salute senza stress.
Riferimenti
Adam, Tanja C, et al. "Il cortisolo è associato negativamente alla sensibilità all'insulina nei giovani latini in sovrappeso". Il Journal of Clinical Endocrinology and Metabolism, The Endocrine Society, ottobre 2010, www.ncbi.nlm.nih.gov/pmc/articles/PMC3050109/.
De Feo, P, et al. "Contributo del cortisolo alla controregolazione del glucosio negli esseri umani". L'American Journal of Physiology, Biblioteca nazionale di medicina degli Stati Uniti, luglio 1989, pubmed.ncbi.nlm.nih.gov/2665516/.
Hucklebridge, FH, et al. "Il risveglio della risposta al cortisolo e i livelli di glucosio nel sangue". Life Sciences, Biblioteca nazionale di medicina degli Stati Uniti, 1999, pubmed.ncbi.nlm.nih.gov/10201642/.
Joseph, Joshua J e Sherita H Golden. "Disregolazione del cortisolo: il legame bidirezionale tra stress, depressione e diabete mellito di tipo 2". Annali della New York Academy of Sciences, Biblioteca nazionale di medicina degli Stati Uniti, marzo 2017, www.ncbi.nlm.nih.gov/pmc/articles/PMC5334212/.
Kamba, Aya, et al. "Associazione tra livelli di cortisolo sierico più elevati e diminuzione della secrezione di insulina in una popolazione generale". PloS One, Biblioteca Pubblica della Scienza, 18 nov. 2016, www.ncbi.nlm.nih.gov/pmc/articles/PMC5115704/.
Lee, Do Yup, et al. "Aspetti tecnici e clinici del cortisolo come marker biochimico di stress cronico". Rapporti BMB, Società coreana di biochimica e biologia molecolare, aprile 2015, www.ncbi.nlm.nih.gov/pmc/articles/PMC4436856/.
Thau, Lauren, et al. "Fisiologia, cortisolo". In: StatPearls [Internet]. L'isola del tesoro (FL), StatPearls Publishing, 6 settembre 2021, www.ncbi.nlm.nih.gov/books/NBK538239.
Il corpo è una macchina ben funzionante che può sopportare tutto ciò che viene lanciato sulla sua strada. Tuttavia, quando subisce un infortunio, il processo di guarigione naturale del corpo farà sì che il corpo possa tornare alle sue attività quotidiane. Il processo di guarigione di un muscolo ferito varia in tutto il corpo. A seconda della gravità del danno e della durata del processo di guarigione, il corpo può riprendersi da pochi giorni a pochi mesi. Uno dei processi di guarigione più estenuanti che il corpo deve sopportare è la rottura del tendine calcaneare.
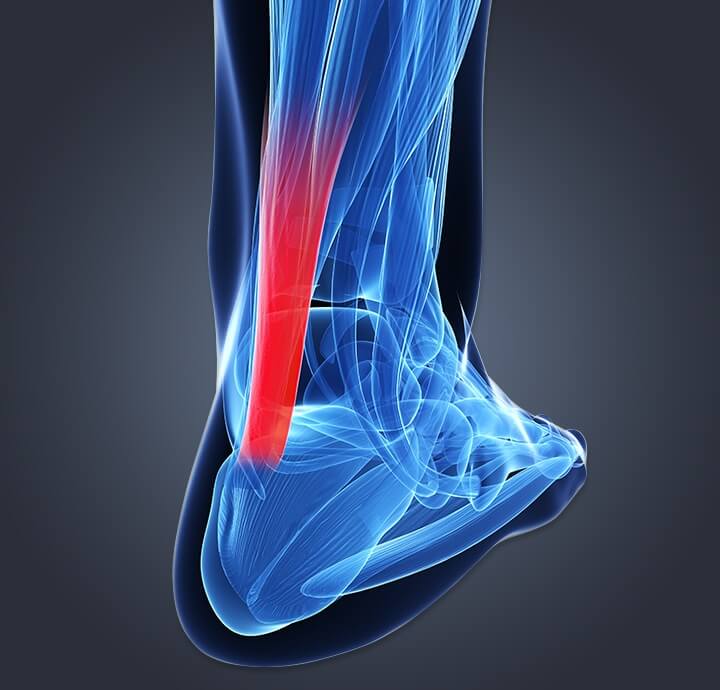
Il tendine calcaneare
Il tendine calcaneare o tendine di Achille è un tendine spesso che si trova nella parte posteriore della gamba. Questo muscolo-tendine è ciò che fa muovere il corpo mentre si cammina, si corre o si salta. Non solo, il tendine calcaneare è il tendine più forte del corpo e collega i muscoli gastrocnemio e soleo all'osso del tallone. Quando il tendine calcaneare si rompe, il processo di guarigione può durare da settimane a mesi fino a quando non è completamente guarito.
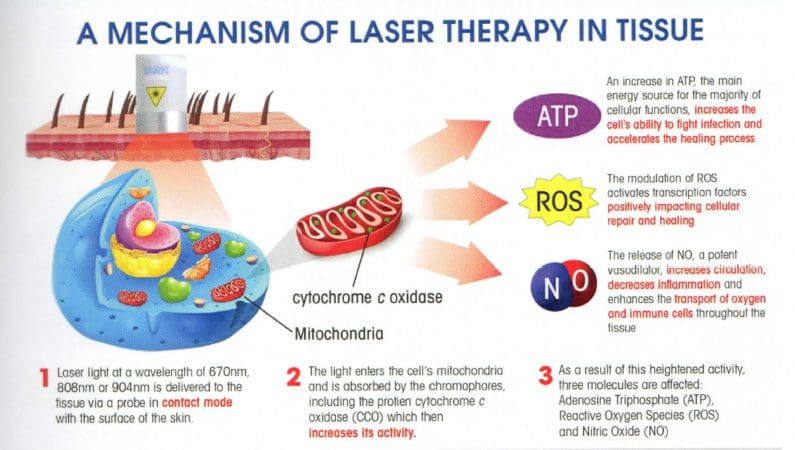
Gli effetti curativi della terapia laser bassa
Uno dei modi che può aiutare il processo di guarigione dei tendini calcaneari danneggiati è la terapia laser a bassa. Studi hanno dimostrato che una bassa terapia laser può accelerare la riparazione del tendine danneggiato dopo una lesione parziale. Non solo quello, ma il pettineInazione degli ultrasuoni e della terapia laser a bassa è stata studiata per essere gli agenti fisici per il trattamento delle lesioni tendinee. Gli studi hanno mostrato che la combinazione di terapia laser a bassa e ultrasuoni ha proprietà benefiche durante il processo di recupero del trattamento delle lesioni del tendine calcaneare.
Lo studio ha trovato che quando i pazienti vengono trattati per i loro tendini calcaneari, i loro livelli di idrossiprolina intorno all'area trattata sono significativamente aumentati con ultrasuoni e laser a bassa tterapia. Le strutture biochimiche e biomeccaniche naturali del corpo sul tendine ferito aumentano, influenzando così il processo di guarigione. Un altro studio ha dimostrato che la terapia laser a bassa può aiutare a ridurre la fibrosi e prevenire lo stress ossidativo nel tendine calcaneare traumatizzato. Lo studio ha anche dimostrato che dopo che il tendine calcaneare è traumatizzato, nell'area interessata si formano infiammazione, angiogenesi, vasodilatazione e matrice extracellulare. Quindi, quando i pazienti vengono trattati con una terapia laser bassa per circa quattordici-ventuno giorni, le loro anomalie istologiche vengono alleviate, riducendo la concentrazione di collagene e la fibrosi; prevenire l'aumento dello stress ossidativo nel corpo.
Conclusione
Nel complesso, si dice che gli effetti della terapia laser a bassa velocità possano aiutare ad accelerare il processo di guarigione della riparazione del tendine calcaneare. I risultati promettenti sono stati dimostrati poiché una bassa terapia laser può aiutare a riparare il tendine danneggiato, riducendo lo stress ossidativo e prevenendo l'escalation della fibrosi, causando più problemi al tendine ferito. E con la combinazione degli ultrasuoni, il tendine calcaneare può riprendersi più velocemente in modo che il corpo possa continuare le sue attività quotidiane senza lesioni prolungate.
Riferimenti:
Demir, Huseyin, et al. "Confronto degli effetti di laser, ultrasuoni e trattamenti combinati laser + ultrasuoni nella guarigione sperimentale dei tendini". Laser in chirurgia e medicina, Biblioteca nazionale di medicina degli Stati Uniti, 2004, pubmed.ncbi.nlm.nih.gov/15278933/.
Fillipin, Lidiane Isabel, et al. "La terapia laser a basso livello (LLLT) previene lo stress ossidativo e riduce la fibrosi nel tendine di Achille traumatizzato dai ratti". Laser in chirurgia e medicina, Biblioteca nazionale di medicina degli Stati Uniti, ottobre 2005, pubmed.ncbi.nlm.nih.gov/16196040/.
Wood, Viviane T, et al. "Modifiche e riallineamento del collagene indotti dalla terapia laser a basso livello e dagli ultrasuoni a bassa intensità nel tendine calcaneare". Laser in chirurgia e medicina, Biblioteca nazionale di medicina degli Stati Uniti, 2010, pubmed.ncbi.nlm.nih.gov/20662033/.
Lo strumento Find A Practitioner di IFM è la più grande rete di riferimento in Medicina Funzionale, creata per aiutare i pazienti a localizzare professionisti di Medicina Funzionale in qualsiasi parte del mondo. I professionisti certificati IFM sono elencati per primi nei risultati di ricerca, data la loro vasta formazione in Medicina Funzionale
Prenotazione online e appuntamenti 24 ore su 7, XNUMX giorni su XNUMX *