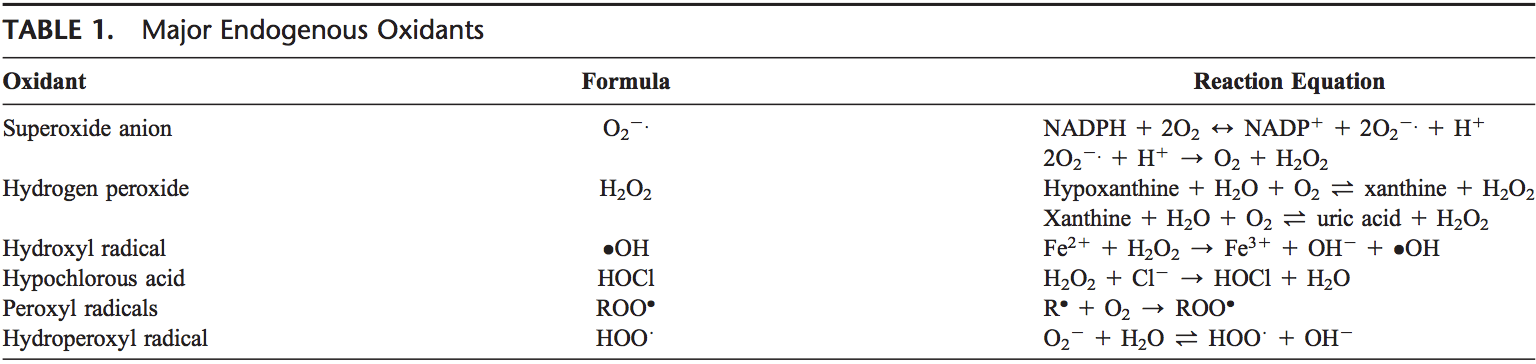
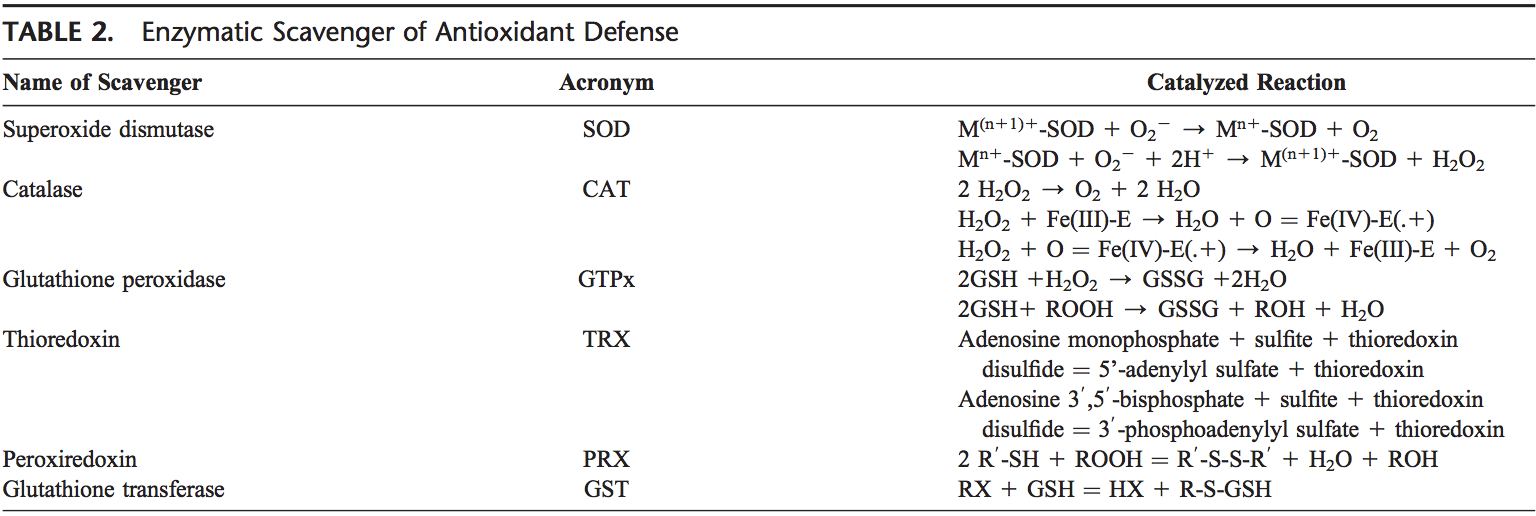
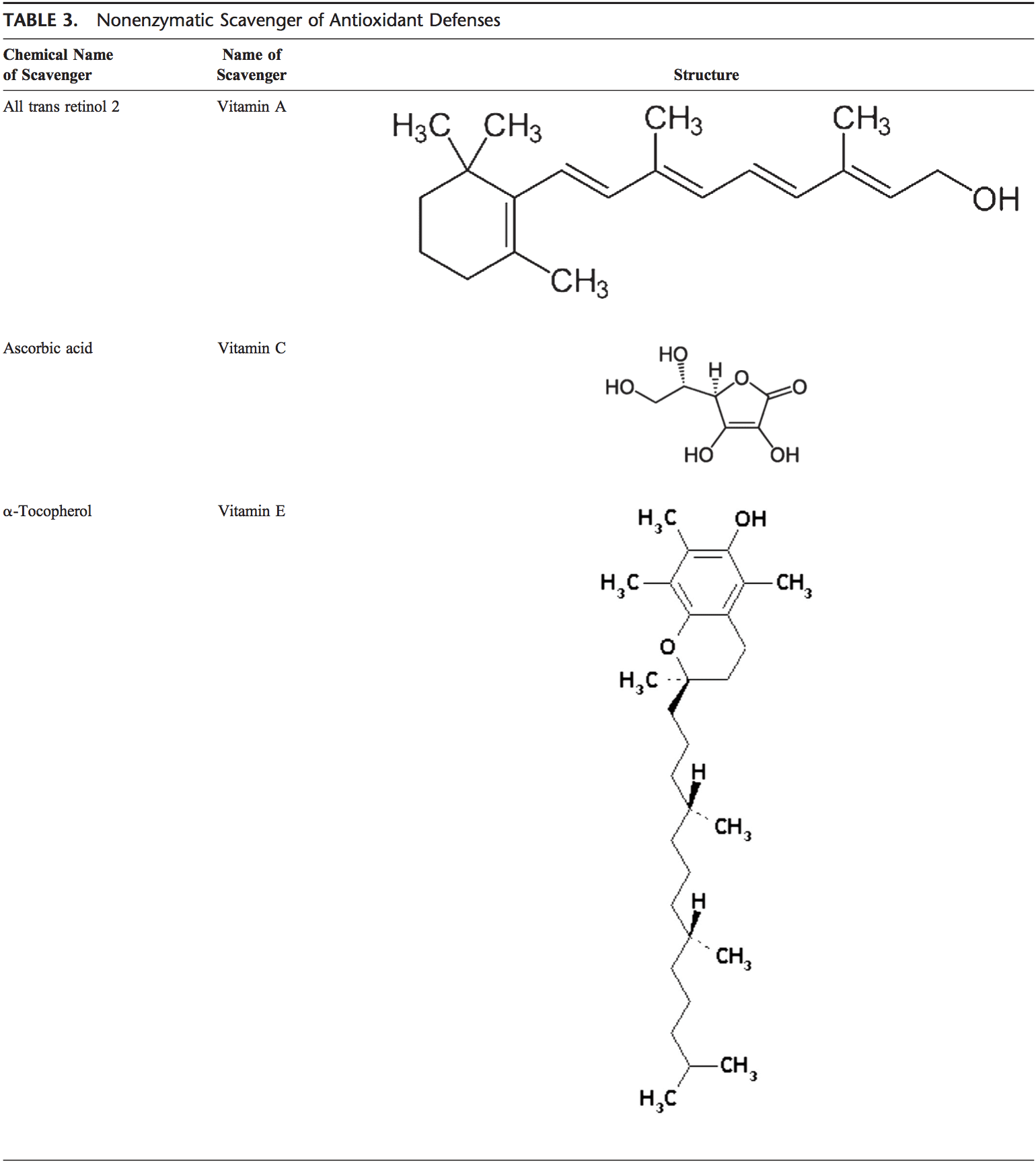
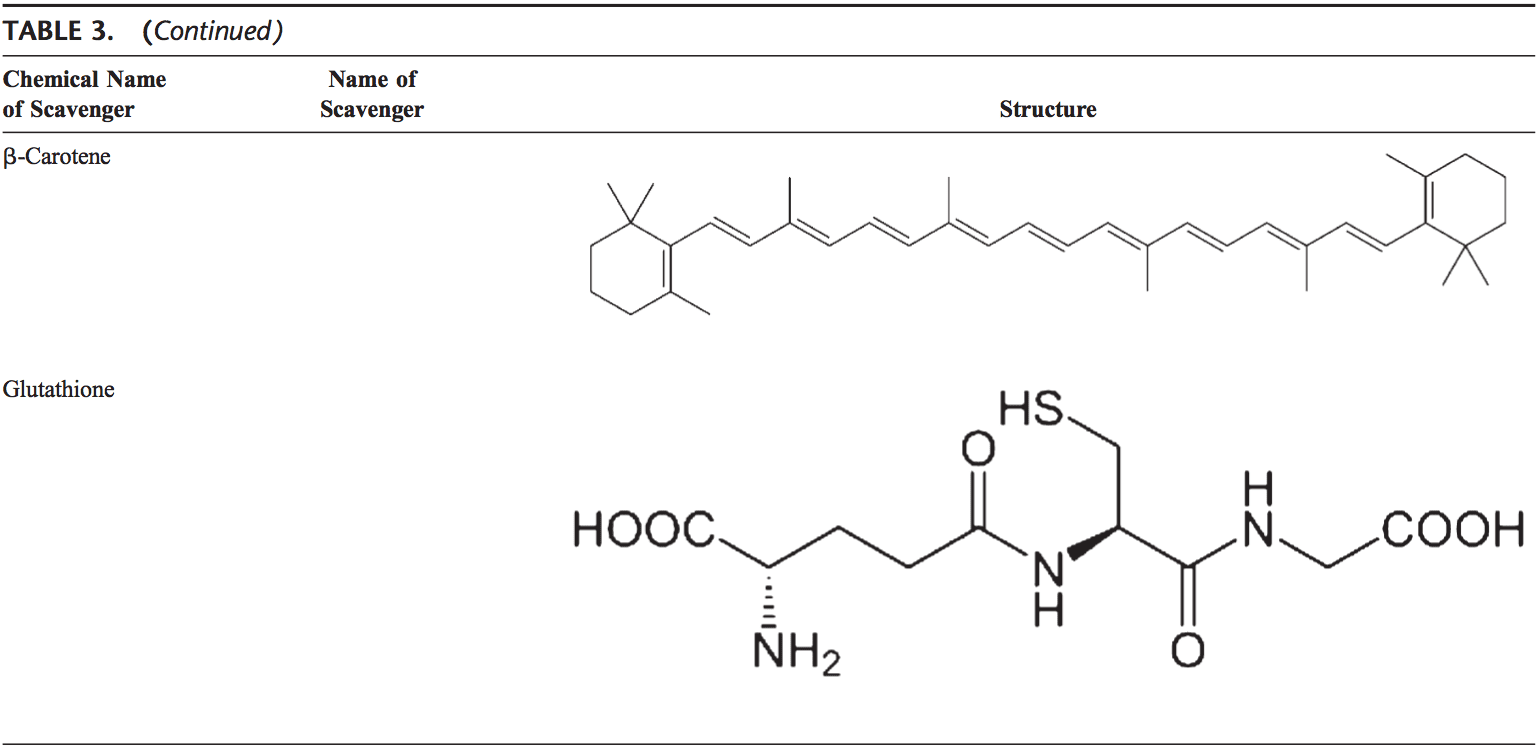
Back Clinic Team di Chiropratica e Medicina Funzionale Stress Ossidativo. Lo stress ossidativo è definito come un disturbo nell'equilibrio tra la produzione di ossigeno reattivo (radicali liberi) e le difese antiossidanti. In altre parole, si tratta di uno squilibrio tra la produzione di radicali liberi e la capacità dell'organismo di contrastare o disintossicare gli effetti nocivi attraverso la neutralizzazione da parte degli antiossidanti. Lo stress ossidativo porta a molte condizioni fisiopatologiche nel corpo. Questi includono malattie neurodegenerative, ad esempio morbo di Parkinson, morbo di Alzheimer, mutazioni genetiche, tumori, sindrome da stanchezza cronica, sindrome dell'X fragile, disturbi cardiaci e dei vasi sanguigni, aterosclerosi, insufficienza cardiaca, infarto e malattie infiammatorie. L'ossidazione avviene in una serie di circostanze:
le cellule usano il glucosio per produrre energia
il sistema immunitario sta combattendo contro i batteri e creando infiammazioni
i corpi detossificano inquinanti, pesticidi e fumo di sigaretta
Ci sono milioni di processi che avvengono nei nostri corpi in un dato momento che possono provocare l'ossidazione. Ecco alcuni sintomi:
stanchezza
Perdita di memoria e / o nebbia cerebrale
Dolore muscolare e articolare
Rughe e capelli grigi
Diminuzione della vista
Mal di testa e sensibilità al rumore
Suscettibilità alle infezioni
Scegliere cibi biologici ed evitare le tossine nel proprio ambiente fa una grande differenza. Questo, insieme alla riduzione dello stress, può essere utile per ridurre l'ossidazione.
Gli ossidanti sono generalmente prodotti in modo controllato al fine di regolare i processi essenziali nel corpo umano, compresa la divisione cellulare, l'infiammazione, la funzione immunitaria, l'autofagia e la risposta allo stress. Tuttavia, la produzione incontrollata di questi ossidanti può contribuire a lo stress ossidativo, che può influire sulla funzione cellulare, portando allo sviluppo di tossicità, malattie croniche e cancro. I meccanismi antiossidanti protettivi del corpo umano sono regolati da una serie di percorsi vitali che controllano la risposta della cellula agli ossidanti. Il fattore nucleare fattore eritroide 2, altrimenti noto come Nrf2, è un regolatore emergente della resistenza cellulare agli ossidanti. Lo scopo di questo articolo è discutere e dimostrare il ruolo emergente di Nrf2 nella funzione mitocondriale.
Astratto
Il fattore di trascrizione NF-E2 fattore 45 correlato a p2 (Nrf2; nome del gene NFE2L2) consente l'adattamento e la sopravvivenza in condizioni di stress regolando l'espressione genica di diverse reti di proteine citoprotettive, inclusi enzimi antiossidanti, antinfiammatori e disintossicanti come proteine che aiutano nella riparazione o nella rimozione delle macromolecole danneggiate. Nrf2 ha un ruolo cruciale nel mantenimento dell'omeostasi redox cellulare regolando la biosintesi, l'utilizzo e la rigenerazione di glutatione, tioredossina e NADPH e controllando la produzione di specie reattive dell'ossigeno da parte dei mitocondri e della NADPH ossidasi. In condizioni omeostatiche, Nrf2 influenza il potenziale della membrana mitocondriale, l'ossidazione degli acidi grassi, la disponibilità di substrati (NADH e FADH2 / succinato) per la respirazione e la sintesi di ATP. In condizioni di stress o stimolazione del fattore di crescita, l'attivazione di Nrf2 contrasta l'aumento della produzione di specie reattive dell'ossigeno nei mitocondri attraverso la sovraregolazione trascrizionale della proteina di disaccoppiamento 3 e influenza la biogenesi mitocondriale mantenendo i livelli del fattore respiratorio nucleare 1 e del recettore attivato dal proliferatore del perossisoma? coattivatore 1, nonché promuovendo la biosintesi dei nucleotidi purinici. Gli attivatori Nrf2 farmacologici, come l'isotiocianato sulforafano presente in natura, inibiscono l'apertura mediata dall'ossidante del poro di transizione della permeabilità mitocondriale e il rigonfiamento mitocondriale. Curiosamente, è stato scoperto che un composto 1,4-difenil-1,2,3-triazolo sintetico, originariamente progettato come attivatore Nrf2, promuove la mitofagia, contribuendo così all'omeostasi mitocondriale complessiva. Pertanto, Nrf2 è un attore di primo piano nel supportare l'integrità strutturale e funzionale dei mitocondri e questo ruolo è particolarmente cruciale in condizioni di stress.
parole chiave:Bioenergetica, Cytoprotection, Keap1, Mitocondri, Nrf2, Radicali liberi
Highlight
Nrf2 ha un ruolo cruciale nel mantenimento dell'omeostasi redox cellulare.
Nrf2 agisce sul potenziale della membrana mitocondriale e sulla sintesi dell'ATP.
Nrf2 influenza l'ossidazione degli acidi grassi mitocondriali.
Nrf2 supporta l'integrità strutturale e funzionale dei mitocondri.
Gli attivatori Nrf2 hanno effetti benefici quando la funzione mitocondriale è compromessa.
Introduzione
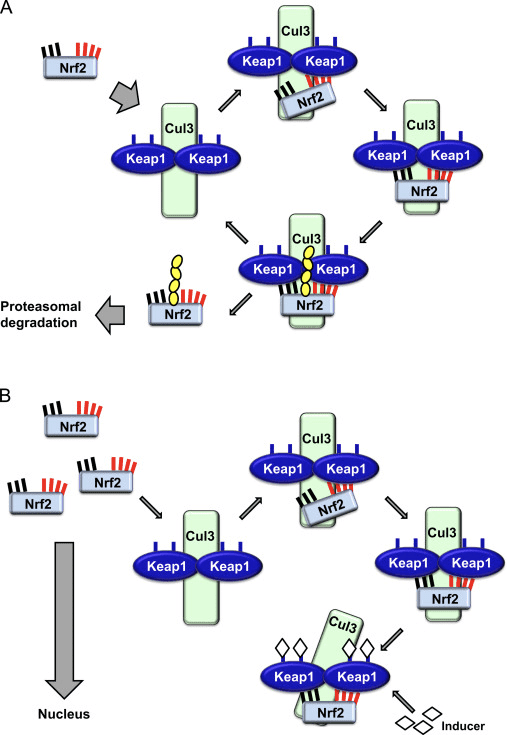
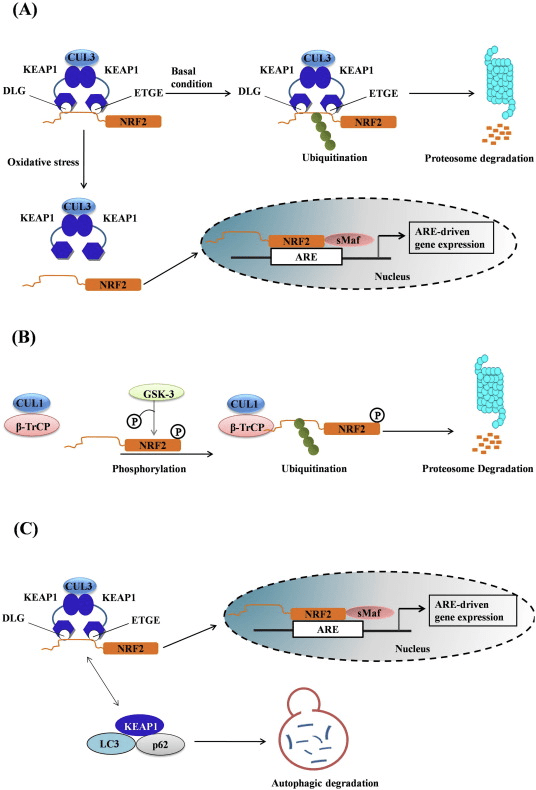
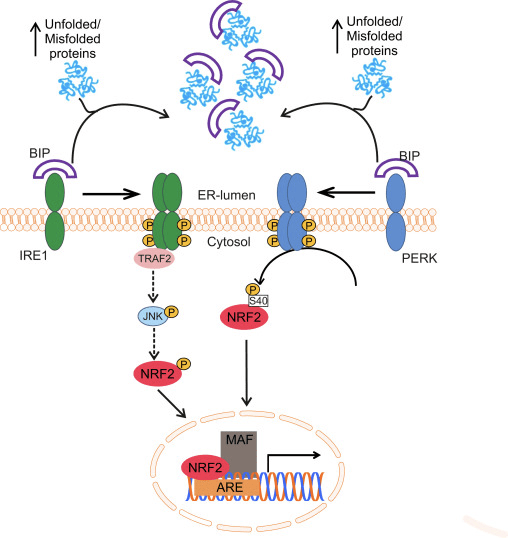
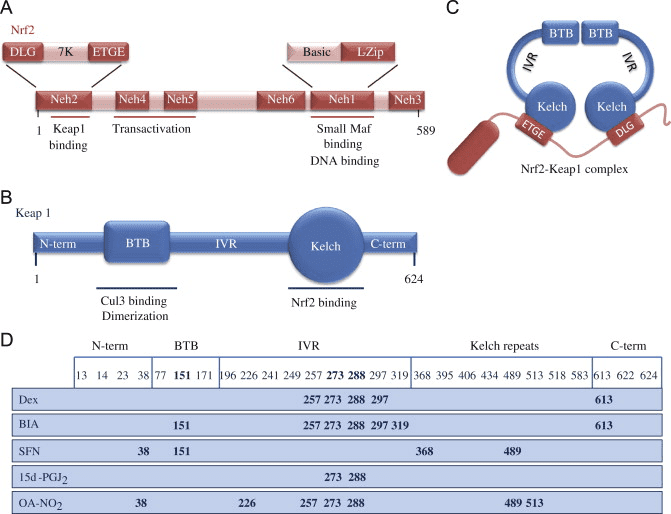
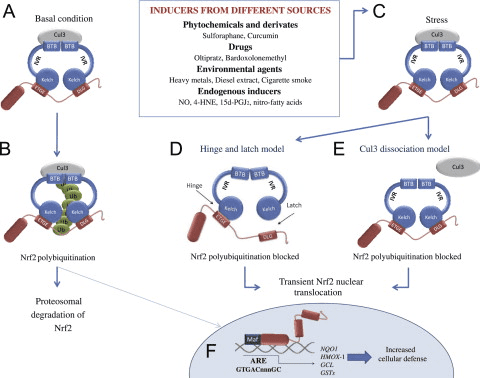
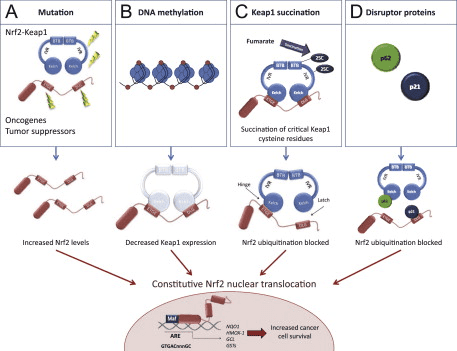
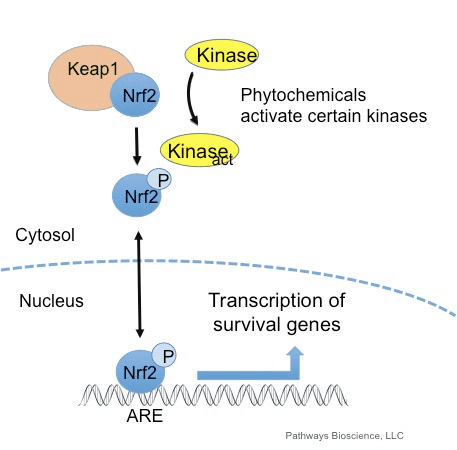
Il fattore di trascrizione NF-E2 fattore 45 correlato a p2 (Nrf2; nome del gene NFE2L2) regola l'espressione di reti di geni che codificano proteine con diverse attività citoprotettive. Nrf2 stesso è controllato principalmente a livello di stabilità proteica. In condizioni basali, Nrf2 è una proteina di breve durata soggetta a continua ubiquitinazione e degradazione proteasomica. Esistono tre sistemi noti di ubiquitina ligasi che contribuiscono alla degradazione di Nrf2. Storicamente, il primo regolatore negativo di Nrf2 scoperto è stata la proteina 1 associata a Kelch-like ECH (Keap1) [1], una proteina adattatore del substrato per Cullin 3 (Cul3) / Rbx1 ubiquitina ligasi [2], [3], [ 4]. Keap1 utilizza un meccanismo ciclico altamente efficiente per indirizzare Nrf2 per l'ubiquitinazione e la degradazione proteasomale, durante la quale Keap1 viene continuamente rigenerato, consentendo al ciclo di procedere (Fig. 1A) [5]. Nrf2 è anche soggetto a degradazione mediata dalla glicogeno sintasi chinasi (GSK) 3 /? - ubiquitina ligasi basata su Cul1 TrCP-dipendente [6], [7]. Più recentemente, è stato riportato che, in condizioni di stress del reticolo endoplasmatico, Nrf2 è ubiquitinato e degradato in un processo mediato dalla E3 ubiquitina ligasi Hrd1 [8].
Figura 1 Il modello ciclico sequenziale di legame e rigenerazione per la degradazione mediata da Keap1 di Nrf2. (A) Nrf2 si lega in sequenza ad un dimero Keap1 gratuito: prima attraverso il suo dominio di legame ETGE (red sticks) ad alta affinità e quindi attraverso il suo dominio di binding DLG (black sticks) a bassa affinità. In questa conformazione del complesso proteico, Nrf2 subisce l'ubiquitinazione ed è mirato alla degradazione del proteasoma. Free Keap1 è rigenerato e in grado di legarsi a Nrf2 nuovamente tradotto e il ciclo ricomincia. (B) Gli induttori (diamanti bianchi) reagiscono con le cisteine del sensore di Keap1 (bastoncini blu), portando ad un cambiamento conformazionale e ad una alterata attività dell'adattatore del substrato. Free Keap1 non viene rigenerato, e il nuovo Nrf2 sintetizzato si accumula e si trasporta nel nucleo.
Oltre a servire come proteina dell'adattatore del substrato della ubiquitina ligasi, Keap1 è anche il sensore per un'ampia gamma di attivatori di piccole molecole di Nrf2 (denominati induttori) [9]. Gli induttori bloccano il ciclo di degradazione mediata da Keap1 di Nrf2 modificando chimicamente residui di cisteina specifici all'interno di Keap1 [10], [11] o interrompendo direttamente l'interfaccia di associazione KfXXUMX: Nrf1 [2], [12]. Di conseguenza, Nrf13 non viene degradato e il fattore di trascrizione si accumula e si traslocano nel nucleo (Figura 2B), dove forma un eterodimero con una piccola proteina Maf; si lega agli elementi di risposta antiossidante, le regioni regolatorie a monte dei suoi geni bersaglio; e avvia la trascrizione [1], [14], [15]. La batteria di bersagli Nrf16 comprende proteine con diverse funzioni citoprotettive, compresi enzimi del metabolismo xenobiotico, proteine con funzioni antiossidanti e antinfiammatorie e subunità proteasomali, così come proteine che regolano l'omeostasi redox cellulare e partecipano al metabolismo intermedio.
Nrf2: un Master Regulator of Cellular Redox Homeostasis
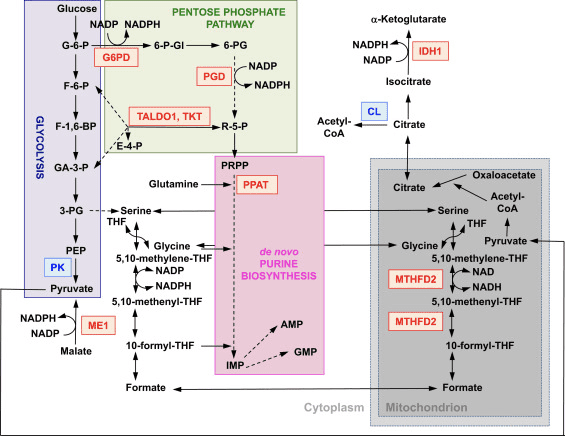
La funzione di Nrf2 come regolatore principale dell'omeostasi redox cellulare è ampiamente riconosciuta. L'espressione genica sia della subunità catalitica che di quella regolatrice della? -Glutamil cisteina ligasi, l'enzima che catalizza la fase di limitazione della velocità nella biosintesi del glutatione ridotto (GSH), è regolata direttamente da Nrf2 [17]. La subunità xCT del sistema xc-, che importa la cistina nelle cellule, è anche un bersaglio trascrizionale diretto di Nrf2 [18]. Nella cellula, la cistina subisce la conversione in cisteina, un precursore della biosintesi del GSH. Oltre al suo ruolo nella biosintesi del GSH, Nrf2 fornisce i mezzi per il mantenimento del glutatione nel suo stato ridotto mediante la regolazione trascrizionale coordinata della glutatione reduttasi 1 [19], [20], che riduce il glutatione ossidato a GSH utilizzando equivalenti riducenti da NADPH . Il NADPH richiesto è fornito da quattro principali enzimi che generano NADPH, enzima malico 1 (ME1), isocitrato deidrogenasi 1 (IDH1), glucosio-6-fosfato deidrogenasi (G6PD) e 6-fosfogluconato deidrogenasi (PGD), che sono trascrizionalmente regolato in parte da Nrf2 (Fig. 2) [21], [22], [23], [24]. Curiosamente, Nrf2 regola anche l'espressione genica inducibile delle forme citosolica, microsomiale e mitocondriale dell'aldeide deidrogenasi [25], che utilizzano NAD (P) + come cofattore, dando origine a NAD (P) H. In effetti, i livelli di NADPH e il rapporto NADPH / NADP + sono inferiori nei fibroblasti embrionali isolati da topi Nrf2-knockout (Nrf2-KO) rispetto alle cellule delle loro controparti wild-type (WT), e i livelli di NADPH diminuiscono con l'abbattimento di Nrf2 in linee cellulari cancerose con Nrf2 costitutivamente attivo [26]. Come previsto, i livelli di GSH sono inferiori nelle cellule in cui Nrf2 è stato interrotto; al contrario, l'attivazione di Nrf2 per via genetica o farmacologica porta alla sovraregolazione del GSH [27], [28], [29]. È importante sottolineare che Nrf2 regola anche l'espressione genica della tioredossina [30], [31], [32], tioredossina reduttasi 1 [28], [29], [32], [33] e sulfiredossina [34], che sono essenziali per la riduzione dei tioli proteici ossidati.
Figura 2 Il ruolo di Nrf2 nel metabolismo delle cellule in rapida proliferazione. Nrf2 è un regolatore positivo dei geni che codificano per gli enzimi sia nel braccio ossidativo [cioè, glucosio-6-fosfato deidrogenasi (G6PD) e 6-fosfogluconato deidrogenasi (PGD)] che nel braccio non ossidativo [cioè, transaldolasi 1 (TALDO1) e transketolasi ( TKT)] della via del pentoso fosfato. G6PD e PGD generano NADPH. Nrf2 regola anche l'espressione genica degli altri due enzimi che generano NADPH, l'enzima malico 1 (ME1) e l'isocitrato deidrogenasi 1 (IDH1). L'espressione genica della fosforibosil pirofosfato amidotransferasi (PPAT), che catalizza l'ingresso nella via biosintetica delle purine de novo, è regolata positivamente anche da Nrf2, così come l'espressione della metilenetetraidrofolato deidrogenasi 2 (MTHFD2), un mitocondriale con un ruolo enzimatico critico fornendo unità a un carbonio per la biosintesi delle purine de novo. La piruvato chinasi (PK) è regolata negativamente da Nrf2 e si prevede che favorisca l'accumulo di intermedi glicolitici e, insieme a G6PD, la canalizzazione dei metaboliti attraverso la via del pentoso fosfato e la sintesi di acidi nucleici, amminoacidi e fosfolipidi. Nrf2 regola negativamente l'espressione genica dell'ATP-citrato liasi (CL), che può aumentare la disponibilità di citrato per l'utilizzo mitocondriale o (tramite isocitrato) per IDH1. Il rosso e il blu indicano rispettivamente la regolazione positiva e negativa. Il mitocondrio è mostrato in grigio. Abbreviazioni dei metaboliti: G-6-P, glucosio 6-fosfato; F-6-P, fruttosio 6-fosfato; F-1,6-BP, fruttosio 1,6-bisfosfato; GA-3-P, gliceraldeide 3-fosfato; 3-PG, 3-fosfoglicerato; PEP, fosfoenolpiruvato; 6-P-Gl, 6-fosfogluconolattone; 6-PG, 6-fosfogluconato; R-5-P, ribulosio 5-fosfato; PRPP, 5-fosforibosil -? - 1-pirofosfato; THF, tetraidrofolato; IMP, inosina monofosfato; AMP, adenosina monofosfato; GMP, guanosina monofosfato.
Dato il ruolo cruciale di Nrf2 come regolatore principale dell'omeostasi redox cellulare, non è sorprendente che, rispetto alle cellule WT, i livelli di specie reattive dell'ossigeno (ROS) siano più alti nelle cellule in cui Nrf2 è stato interrotto (Nrf2-KO) [35]. Questa differenza è particolarmente evidente in caso di sfida con agenti che causano stress ossidativo. Inoltre, le cellule carenti di Nrf2 sono molto più sensibili alla tossicità degli ossidanti di vario tipo e non possono essere protette dagli induttori Nrf2 che, nelle stesse condizioni, forniscono una protezione efficiente e duratura alle cellule WT [29], [36] , [37]. Oltre all'omeostasi della redox cellulare, Nrf2 è anche fondamentale per il mantenimento dell'omeostasi redox mitocondriale. Pertanto, rispetto al WT, il pool totale di NADH mitocondriale è significativamente aumentato in Keap1-KO e drasticamente diminuito nelle cellule Nrf2-KO [35].
Utilizzando l'imaging di cellule vive, abbiamo recentemente monitorato i tassi di produzione di ROS in coclee glioneuronali primarie e fette di tessuto cerebrale isolate da topi WT, Nrf2-KO o Keap1-knockdown (Keap1-KD) [38]. Come previsto, il tasso di produzione di ROS era più veloce nelle cellule e nei tessuti Nrf2-KO rispetto alle controparti WT. Tuttavia, abbiamo fatto l'osservazione inaspettata che, rispetto al WT, le cellule Keap1-KD hanno anche tassi più alti di produzione di ROS, sebbene l'entità della differenza tra i genotipi di WT e Keap1-KD fosse inferiore a quella tra WT e Nrf2-KO . Abbiamo quindi analizzato i livelli di mRNA di NOX2 e NOX4, le subunità catalitiche delle due isoforme di NADPH ossidasi (NOX) che sono state implicate nella patologia cerebrale e hanno scoperto che NOX2 è drasticamente aumentato in condizioni di deficit di Nrf2, mentre NOX4 è sovraregolato quando Nrf2 è costitutivamente attivato, anche se in misura minore. Quantitativamente, l'entità della sovraregolazione nelle cellule e nei tessuti dai topi mutanti è parallela ai corrispondenti aumenti della produzione di ROS [38]. È interessante notare che Nrf2 non solo regola la NADPH ossidasi, ma il ROS prodotto dalla NADPH ossidasi può attivare Nrf2, come mostrato nelle cellule epiteliali polmonari e nei cardiomiociti [39], [40]. Inoltre, uno studio molto recente ha dimostrato che l'attivazione NADPH ossidasi-dipendente di Nrf2 costituisce un importante meccanismo endogeno per la protezione contro il danno mitocondriale e la morte cellulare nel cuore durante il sovraccarico di pressione cronica [41].
Oltre all'attività catalitica della NADPH ossidasi, la respirazione mitocondriale è un'altra importante fonte intracellulare di ROS. Con l'uso della sonda MitoSOX specifica dei mitocondri, abbiamo esaminato il contributo dei ROS dell'origine mitocondriale alla produzione complessiva di ROS in colture primarie glioneuronali isolate da topi WT, Nrf2-KO o Keap1-KD [38]. Come previsto, le cellule Nrf2-KO presentavano tassi più elevati di produzione di ROS mitocondriale rispetto al WT. In accordo con i risultati della produzione complessiva di ROS, i tassi di produzione di ROS dei mitocondri in Keap1-KD erano anche più alti rispetto alle cellule WT. È importante sottolineare che il blocco del complesso I con rotenone ha causato un notevole aumento della produzione di ROS mitocondriale in entrambe le cellule WT e Keap1-KD, ma non ha avuto alcun effetto sulle cellule Nrf2-KO. In contrasto con l'aumento previsto della produzione di ROS mitocondriale nelle cellule WT dopo l'aggiunta di piruvato (per aumentare la disponibilità di NADH, aumentare il potenziale di membrana mitocondriale e normalizzare la respirazione), la produzione di ROS è diminuita nelle cellule Nrf2-KO. Insieme, questi risultati suggeriscono fortemente che, in assenza di Nrf2: (i) l'attività del complesso I è compromessa, (ii) l'attività compromessa del complesso I è dovuta alla limitazione dei substrati e (iii) l'attività compromessa del complesso I è una delle ragioni principali per l'aumento della produzione di ROS mitocondriale, probabilmente a causa del flusso di elettroni inversi dal complesso II.
Nrf2 Influisce sul potenziale e sulla respirazione della membrana mitocondriale
Il potenziale di membrana mitocondriale (?? m) è un indicatore universale della salute mitocondriale e dello stato metabolico della cellula. In una cellula sana, ?? m è mantenuto dalla catena respiratoria mitocondriale. È interessante notare che un'etichettatura isotopica stabile con aminoacidi in uno studio di proteomica basato su colture nella linea cellulare MCF10A epiteliale mammaria non tumorale negativa al recettore degli estrogeni ha dimostrato che il componente della catena di trasporto degli elettroni mitocondriale NDUFA4 è sovraregolato dall'attivazione farmacologica (da sulforafano) di Nrf2, mentre la sovraregolazione genetica di Nrf2 (da Keap1 knockdown) porta alla sottoregolazione delle subunità della citocromo c ossidasi COX2 e COX4I1 [42]. Uno studio del proteoma epatico utilizzando l'elettroforesi su gel bidimensionale e la spettrometria di massa di desorbimento / ionizzazione laser assistita da matrice ha scoperto che Nrf2 regola l'espressione della subunità ATP sintasi? [43]. Inoltre, è stato riportato che la proteina mitocondriale DJ-1, che svolge un ruolo nel mantenimento dell'attività del complesso I [44], stabilizza Nrf2 [45], [46], sebbene gli effetti neuroprotettivi dell'attivazione farmacologica o genetica di Nrf2 sono indipendenti da DJ-1 [47]. Tuttavia, le conseguenze di queste osservazioni per la funzione mitocondriale non sono state studiate.
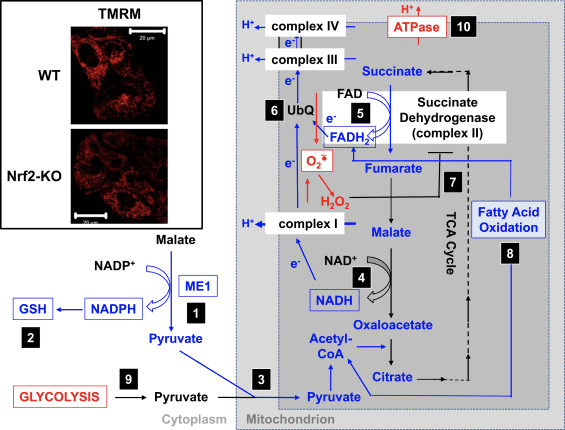
In accordo con l'attività ridotta del complesso I in condizioni di deficit di Nrf2, il basale ?? m è inferiore nei fibroblasti embrionali di topo Nrf2-KO (MEF) e nelle cellule glioneuronali primarie coltivate rispetto alle loro controparti WT (Fig.3, riquadro) [35]. Al contrario, il basale ?? m è più alto quando Nrf2 è geneticamente costitutivamente sovraregolato (per knockdown o knockout di Keap1). Queste differenze di m tra i genotipi indicano che la respirazione è influenzata dall'attività di Nrf2. Infatti, la valutazione del consumo di ossigeno allo stato basale ha rivelato che, rispetto al WT, il consumo di ossigeno è inferiore nei MEF Nrf2-KO e Keap1-KO, rispettivamente di ~ 50 e ~ 35%.
Figura 3 Meccanismo proposto per la funzione mitocondriale compromessa in condizioni di deficit di Nrf2. (1) I livelli ridotti di ME1, IDH1, G6PD e PGD determinano livelli di NADPH inferiori. (2) Anche i livelli di GSH sono bassi. (3) La bassa attività di ME1 può diminuire il pool di piruvato che entra nei mitocondri. (4) La generazione di NADH è più lenta, determinando una ridotta attività del complesso I e una maggiore produzione di ROS mitocondriali. (5) Anche la riduzione di FAD a FADH2 nelle proteine mitocondriali è diminuita, abbassando il flusso di elettroni da FADH2 a UbQ e nel complesso III. (6) La formazione più lenta di UbQH2 può ridurre l'attività enzimatica della succinato deidrogenasi. (7) I livelli aumentati di ROS possono inibire ulteriormente l'attività del complesso II. (8) La minore efficienza dell'ossidazione degli acidi grassi contribuisce alla ridotta disponibilità di substrato per la respirazione mitocondriale. (9) La glicolisi è potenziata come meccanismo compensatorio per la ridotta produzione di ATP nella fosforilazione ossidativa. (10) L'ATP sintasi opera al contrario per mantenere ?? m. Il rosso e il blu indicano rispettivamente la sovraregolazione e la sottoregolazione. Le caselle indicano la disponibilità di prove sperimentali. L'inserto mostra immagini dei mitocondri degli astrociti corticali WT e Nrf2-KO visualizzati dalla sonda fluorescente potenziometrica tetrametilrodamina metilestere (TMRM; 25 nM). Barra della scala, 20 m.
Queste differenze di m e respirazione tra i genotipi si riflettono nella velocità di utilizzo dei substrati per la respirazione mitocondriale. L'applicazione di substrati per il ciclo dell'acido tricarbossilico (TCA) (malato / piruvato, che a sua volta aumenta la produzione del substrato del complesso I NADH) o del metil succinato, un substrato per il complesso II, provoca un aumento graduale di ?? m in entrambi i pesi e neuroni Keap1-KD, ma il tasso di aumento è maggiore nelle cellule Keap1-KD. Ancora più importante, le forme della risposta a questi substrati del ciclo TCA sono diverse tra i due genotipi, per cui il rapido aumento di ?? m nelle cellule Keap1-KD dopo l'aggiunta del substrato è seguito da un rapido calo piuttosto che da un plateau, suggerendo un insolitamente rapido consumo di substrato. Questi risultati sono in stretto accordo con i livelli molto più bassi (del 50-70%) di malato, piruvato e succinato che sono stati osservati dopo un impulso di 1 ora di glucosio [U-13C6] in Keap1-KO rispetto a WT MEF cellule [24]. Nei neuroni Nrf2-KO, solo il piruvato è in grado di aumentare il ?? m, mentre il malato e il metil succinato causano una lieve depolarizzazione. L'effetto di Nrf2 sulla produzione di substrato mitocondriale sembra essere il meccanismo principale con cui Nrf2 influenza la funzione mitocondriale. L'indice di redox NADH mitocondriale (l'equilibrio tra il consumo di NADH da parte del complesso I e la produzione di NADPH nel ciclo TCA) è significativamente inferiore nelle cellule Nrf2-KO rispetto alle loro controparti WT, e inoltre, i tassi di rigenerazione dei pool di NADH e FADH2 dopo l'inibizione del complesso IV (mediante l'uso di NaCN) sono più lenti nelle cellule mutanti.
Nei mitocondri isolati dal cervello e dal fegato murini, l'integrazione di substrati per il complesso I o per il complesso II aumenta il tasso di consumo di ossigeno più fortemente quando Nrf2 è attivato e meno efficientemente quando Nrf2 viene interrotto [35]. Pertanto, il malato induce un tasso più elevato di consumo di ossigeno in Keap1-KD rispetto a WT, ma il suo effetto è più debole nei mitocondri Nrf2-KO. Allo stesso modo, in presenza di rotenone (quando il complesso I è inibito), il succinato attiva il consumo di ossigeno in misura maggiore in Keap1-KD rispetto a WT, mentre la risposta nei mitocondri Nrf2-KO è diminuita. Inoltre, le colture neuronali primarie Nrf2-KO e i topi sono più sensibili alla tossicità degli inibitori del complesso II acido 3-nitropropionico e malonato, mentre il trapianto intrastriatale di astrociti con sovraesprimono Nrf2 è protettivo [48], [49]. Allo stesso modo, i topi Nrf2-KO sono più sensibili, mentre l'attivazione genetica o farmacologica di Nrf2 ha effetti protettivi contro la neurotossicità causata dallo ione 1-metil-4-fenilpiridinio del complesso I inibitore nell'1-metil-4-fenil-1,2,3,6, 49-tetraidropiridina modello animale del morbo di Parkinson [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [XNUMX].
Il rapporto di controllo respiratorio (RCR), il rapporto tra la respirazione di Stato 3 (stimolata da ADP) e di Stato 4 (nessun ADP presente), è diminuito in assenza di Nrf2, ma l'RCR è simile tra i mitocondri Keap1-KD e WT [35 ]. Poiché l'RCR è un'indicazione del grado di accoppiamento dell'attività della catena respiratoria mitocondriale alla fosforilazione ossidativa, questo risultato indica che il tasso più elevato di respirazione nei mitocondri Keap1-KD non è dovuto al disaccoppiamento della fosforilazione ossidativa. Suggerisce inoltre che la fosforilazione ossidativa è più efficiente quando Nrf2 è attivato. Il più alto tasso di respirazione nei mitocondri Keap1-KD è coerente con i livelli più alti di produzione di ROS mitocondriali [38] poiché tassi di respirazione più alti possono portare a una maggiore perdita di elettroni. Tuttavia, in condizioni di stress ossidativo, l'aumento della produzione di ROS è contrastato dalla sovraregolazione trascrizionale dipendente da Nrf2 della proteina di disaccoppiamento 3 (UCP3), che aumenta la conduttanza protonica della membrana interna mitocondriale e di conseguenza diminuisce la produzione di superossido [62]. Molto recentemente, è stato dimostrato che il prodotto di perossidazione lipidica 4-idrossi-2-nonenale media la sovraregolazione Nrf2-dipendente di UCP3 nei cardiomiociti; questo potrebbe essere particolarmente importante per la protezione in condizioni di stress ossidativo come quelle durante la riperfusione di ischemia [63].
Nrf2 influenza l'efficienza della fosforilazione ossidativa e la sintesi dell'ATP
In accordo con l'effetto di Nrf2 sulla respirazione, nei mitocondri del cervello e del fegato, la carenza di Nrf2 si traduce in una diminuzione dell'efficienza della fosforilazione ossidativa (come stimato dal rapporto tra ADP e ossigeno, che viene consumato per la sintesi di ATP), mentre l'attivazione di Nrf2 (Keap1 -KD) ha l'effetto opposto [35]. Rispetto al WT, i livelli di ATP sono significativamente più alti nelle cellule con sovraregolazione costitutiva di Nrf2 e inferiori quando Nrf2 viene abbattuto [64] o interrotto [35]. Inoltre, l'uso di inibitori della fosforilazione ossidativa (oligomicina) o della glicolisi (acido iodoacetico) ha rivelato che Nrf2 cambia il modo in cui le cellule producono ATP. Pertanto, nei neuroni WT, l'oligomicina causa un calo completo dell'ATP e l'acido iodoacetico non ha ulteriori effetti. Sorprendentemente, nelle cellule Nrf2-KO, l'oligomicina aumenta i livelli di ATP, che vengono poi lentamente, ma completamente, impoveriti dall'acido iodoacetico, indicando che in assenza di Nrf2, la glicolisi e non la fosforilazione ossidativa è la principale fonte di produzione di ATP. È interessante notare che, nonostante la maggiore efficienza della fosforilazione ossidativa nelle cellule Keap1-KD, l'aggiunta di oligomicina provoca una diminuzione dell'80% circa dei livelli di ATP e l'acido iodoacetico provoca un'ulteriore diminuzione del 20% circa. Pertanto, la carenza di Nrf2 o la sua attivazione costitutiva riduce il contributo della fosforilazione ossidativa e aumenta il contributo della glicolisi alla sintesi dell'ATP. Questo effetto è particolarmente pronunciato quando Nrf2 è assente ed è coerente con la dipendenza del ?? m dalla presenza di glucosio nel mezzo [35] e l'aumento dei livelli di intermedi glicolitici (G-6-P, F-6-P , diidrossiacetone fosfato, piruvato e lattato) dopo il knockdown di Nrf2 [24].
L'aumento dei livelli di ATP dopo l'inibizione dell'F1F0-ATPasi da parte dell'oligomicina indica che in assenza di Nrf2, l'F1F0-ATPasi funziona come ATPasi e non ATP sintasi, cioè opera al contrario. Tale inversione di attività molto probabilmente riflette la necessità di pompare protoni attraverso la membrana mitocondriale interna nel tentativo di mantenere il ?? m, che è cruciale per l'integrità funzionale di questo organello. L'inversione della funzione della F1F0-ATPasi è evidenziata anche dalla depolarizzazione mitocondriale osservata dopo somministrazione di oligomicina alle cellule Nrf2-KO, che è in netto contrasto con l'iperpolarizzazione che si verifica nelle loro controparti carenti di WT o Keap1 [35]. Nel complesso, sembra che in condizioni di carenza di Nrf2 l'ATP sia prodotto principalmente nella glicolisi, e questo ATP viene poi utilizzato in parte dalla F1F0-ATPasi per mantenere ?? m.
Nrf2 migliora l'ossidazione degli acidi grassi mitocondriali
L'effetto del deficit di Nrf2 sul ?? m è particolarmente pronunciato quando le cellule vengono incubate in un mezzo senza glucosio, e il ?? m è inferiore del ~ 50% in Nrf2-KO rispetto alle cellule WT [35]. In condizioni di privazione del glucosio, l'ossidazione degli acidi grassi mitocondriali (FAO) è un importante fornitore di substrati per la respirazione e la fosforilazione ossidativa, suggerendo che Nrf2 può influenzare la FAO. In effetti, l'efficienza della FAO sia per l'acido palmitico saturo a catena lunga (C16: 0) che per l'acido esanoico a catena corta (C6: 0) è maggiore nei MEF Keap1-KO e nei mitocondri isolati del cuore e del fegato rispetto ai loro Controparti WT, mentre è inferiore nelle cellule Nrf2-KO e nei mitocondri [65]. Questi effetti sono anche molto rilevanti per l'uomo: infatti, negli studi di intervento sull'uomo con diete ricche di glucorafanina, il precursore del classico attivatore Nrf2 sulforafano, sono stati segnalati cambiamenti metabolici indicativi di una migliore integrazione della FAO con l'attività del ciclo TCA [ 66].
Durante la prima fase della FAO mitocondriale, l'idrogeno pro-R del? -Carbonio lascia come idruro che riduce il cofattore FAD a FADH2, che a sua volta trasferisce gli elettroni all'ubiquinone (UbQ) nella catena respiratoria, contribuendo in ultima analisi alla produzione di ATP . Mentre la stimolazione della FAO da parte della palmitoilcarnitina in assenza di glucosio causa il previsto aumento dei livelli di ATP nelle cellule WT e Keap1-KO, con l'aumento dell'ATP più veloce nelle cellule Keap1-KO, lo stesso trattamento non produce cambiamenti di ATP in Nrf2-KO MEF [65]. Questo esperimento dimostra che, in assenza di Nrf2, la FAO viene soppressa e, inoltre, implica la soppressione della FAO come una delle ragioni per i livelli inferiori di ATP in condizioni di carenza di Nrf2 [35], [64].
In particolare, le cellule T 293 umane in cui Nrf2 è stato silenziato hanno un'espressione inferiore di CPT1 e CPT2 [67], due isoforme di carnitina palmitoiltransferasi (CPT), l'enzima limitante della velocità nella FAO mitocondriale. In accordo, i livelli di mRNA di Cpt1 sono inferiori nei fegati di Nrf2-KO rispetto ai topi WT [68]. CPT catalizza il trasferimento del gruppo acilico di un acile-CoA grasso a catena lunga dal coenzima A alla l-carnitina e consente quindi l'importazione di acilcarnitina dal citoplasma nei mitocondri. Sebbene questo non sia stato ancora esaminato, è possibile che oltre agli effetti di trascrizione sull'espressione di CPT1, Nrf2 possa anche influenzare la funzione di questo enzima controllando i livelli del suo principale inibitore allosterico, il malonil-CoA. Questo perché, tramite un meccanismo che non è chiaro, Nrf2 regola negativamente l'espressione di stearoil CoA desaturasi (SCD) [69] e citrato liasi (CL) [69], [70]. Curiosamente, l'eliminazione diretta o l'inibizione della SCD porta ad un aumento della fosforilazione e attivazione della protein chinasi attivata da AMP (AMPK) [71], [72], [73], e si può ipotizzare che, in assenza di Nrf2, i livelli di SCD aumenterà, a sua volta diminuendo l'attività AMPK. Ciò potrebbe essere ulteriormente aggravato dai livelli proteici ridotti di AMPK che sono stati osservati nei fegati dei topi Nrf2-KO [68], una scoperta che è in stretto accordo con i livelli aumentati di AMPK, che sono stati riportati nei fegati di Keap1-KD topi [74]. Una conseguenza della diminuita attività di AMPK è il sollievo della sua fosforilazione inibitoria (a Ser79) di acetil-CoA carbossilasi (ACC) [75], che potrebbe essere ulteriormente sovraregistrata transcriptionally in assenza di Nrf2 perché è downregolata dall'attivazione di Nrf2 [70 ]. L'elevata attività dell'ACC, in combinazione con l'espressione CL sovraregolata che aumenterà la produzione di acetil-CoA, il substrato per ACC, potrebbe infine aumentare i livelli del prodotto ACC, malonil-CoA. Gli alti livelli di malonil-CoA inibiscono il CPT, riducendo così il trasporto di acidi grassi nei mitocondri. Infine, Nrf2 regola positivamente l'espressione di CD36 [76], una translocasi che importa gli acidi grassi attraverso le membrane plasmatiche e mitocondriali. Quindi, un meccanismo attraverso il quale Nrf2 può influenzare l'efficienza della FAO mitocondriale è regolando l'importazione di acidi grassi a catena lunga nei mitocondri.
Oltre alla regolazione trascrizionale diretta, Nrf2 può anche alterare l'efficienza della FAO mitocondriale con i suoi effetti sul metabolismo del redox cellulare. Questo può essere particolarmente rilevante quando l'attività di Nrf2 è bassa o assente, condizioni che spostano lo stato di ossidoriduzione cellulare verso lo stato ossidato. In effetti, diversi enzimi FAO sono stati identificati come sensibili ai cambiamenti redox. Uno di questi enzimi è l'acil-CoA deidrogenasi a catena molto lunga (VLCAD), che contribuisce più dell'80% all'attività di deidrogenazione del palmitoil-CoA nei tessuti umani [77]. È interessante notare che Hurd et al. [78] hanno dimostrato che VLCAD contiene residui di cisteina che modificano significativamente il loro stato redox dopo l'esposizione di mitocondri di ratto isolati a H2O2. Inoltre, la S-nitrosilazione del VLCAD epatico murino in Cys238 migliora l'efficienza catalitica dell'enzima [79] ed è probabile che l'ossidazione della stessa cisteina possa avere l'effetto opposto, riducendo in definitiva l'efficienza della FAO mitocondriale. È quindi possibile che, sebbene i livelli di espressione di VLCAD non siano significativamente differenti in WT, Nrf2-KO o MEF Keap1-KO [65], l'attività enzimatica di VLCAD potrebbe essere inferiore in assenza di Nrf2 a causa dei livelli più alti di ROS.
Sulla base di tutti questi risultati, si può proporre che (Fig. 3): in assenza di Nrf2, i livelli di NADPH sono inferiori a causa della ridotta espressione di ME1, IDH1, G6PD e PGD. I livelli di glutatione ridotto sono anche inferiori a causa della ridotta espressione di enzimi che partecipano alla sua biosintesi e rigenerazione e ai livelli inferiori di NADPH necessari per la conversione della forma ossidata in glutatione ridotta. La bassa espressione di ME1 diminuirà il pool di piruvato che entra nei mitocondri, con la glicolisi che diventa la principale fonte di piruvato. La generazione di NADH è più lenta, portando ad una ridotta attività del complesso I e ad una maggiore produzione di ROS mitocondriali. Anche la riduzione di FAD a FADH2 è più lenta, almeno in parte a causa di un'ossidazione degli acidi grassi meno efficiente, che compromette il flusso di elettroni da FADH2 a UbQ e nel complesso III. Poiché UbQH2 è un attivatore della succinato deidrogenasi [80], rallentarne la formazione può ridurre l'attività enzimatica della succinato deidrogenasi. I livelli aumentati di superossido e perossido di idrogeno possono inibire ulteriormente l'attività del complesso II [81]. La minore efficienza dell'ossidazione degli acidi grassi contribuisce alla ridotta disponibilità di substrato per la respirazione mitocondriale e alla produzione di ATP nella fosforilazione ossidativa. Come meccanismo compensatorio, la glicolisi è migliorata. L'ATP sintasi funziona al contrario, come ATPasi, nel tentativo di mantenere il ?? m.
Nrf2 e biogenesi mitocondriale
È stato riportato che, rispetto al WT, i fegati dei topi Nrf2-KO hanno un contenuto mitocondriale inferiore (come determinato dal rapporto tra DNA mitocondriale e nucleare); questo è ulteriormente diminuito da un digiuno di 24 ore sia nei topi WT che Nrf2-KO; al contrario, sebbene non sia diverso dal WT in normali condizioni di alimentazione, il contenuto mitocondriale nei topi con elevata attività Nrf2 non è influenzato dal digiuno [82]. È interessante notare che l'integrazione con l'attivatore Nrf2 (R) -? - acido lipoico [83], [84], [85] promuove la biogenesi mitocondriale negli adipociti 3T3-L1 [86]. Due classi di regolatori trascrizionali nucleari svolgono un ruolo critico nella biogenesi mitocondriale. La prima classe sono i fattori di trascrizione, come i fattori respiratori nucleari11 e 2, che controllano l'espressione dei geni che codificano le subunità dei cinque complessi respiratori, i componenti traslazionali mitocondriali e gli enzimi biosintetici dell'eme che sono localizzati nella matrice mitocondriale [88]. Piantadosi et al. [89] hanno dimostrato che la sovraregolazione trascrizionale dipendente da Nrf2 del fattore respiratorio nucleare 1 promuove la biogenesi mitocondriale e protegge dalla citotossicità dell'agente chemioterapico cardiotossico antraciclina doxorubicina. Al contrario, Zhang et al. [82] hanno riferito che l'attivazione genetica di Nrf2 non influenza l'espressione dell'mRNA basale del fattore respiratorio nucleare 1 nel fegato murino.
La seconda classe di regolatori trascrizionali nucleari con funzioni critiche nella biogenesi mitocondriale sono i coattivatori trascrizionali, come il recettore attivato dal proliferatore del perossisoma? coattivatori (PGC) 1? e 1 ?, che interagiscono con i fattori di trascrizione, il meccanismo basale trascrizionale e di splicing dell'RNA e gli enzimi che modificano gli istoni [88], [90], [91]. L'espressione della famiglia di coattivatori PGC1 è influenzata da numerosi segnali ambientali. Il trattamento dei fibroblasti umani con l'attivatore Nrf2 sulforafano provoca un aumento della massa mitocondriale e l'induzione di PGC1? e PGC1? [92], sebbene la potenziale dipendenza da Nrf2 non sia stata esaminata in questo studio. Tuttavia, i topi diabetici in cui Nrf2 è attivato dal knockdown ipomorfico del gene Keap1 (db / db: Keap1flox / ?: Nrf2 + / +) o interrotto (db / db: Keap1flox / ?: Nrf2? /?) Hanno PGC1 epatico inferiore? livelli di espressione rispetto agli animali di controllo (db / db: Keap1flox / +: Nrf2 + / +) [93]. Nessuna differenza nei livelli di mRNA per PGC1? si osservano nel fegato di topi non diabetici che sono WT o Nrf2-KO, mentre questi livelli sono inferiori negli animali con sovraespressione di Nrf2 (Keap1-KD e Keap1-KO specifico per il fegato) [82]. In particolare, un digiuno di 24 ore aumenta i livelli di PGC1? mRNA nel fegato di topi di tutti i genotipi, ma l'aumento è significativamente maggiore nei fegati di Nrf2-KO rispetto ai topi con sovraesprimono WT o Nrf2. Rispetto al WT, i topi Nrf2-KO che presentano un'infezione settica o un danno polmonare acuto a causa di infezione mostrano un'attenuata sovraregolazione trascrizionale del fattore respiratorio nucleare 1 e PGC1? [94], [95]. Insieme, queste osservazioni suggeriscono che il ruolo di Nrf2 nel mantenere i livelli sia del fattore respiratorio nucleare 1 che di PGC1? è complesso e diventa più prominente in condizioni di stress.
Oltre all'espressione di geni che codificano per proteine mitocondriali, la biogenesi mitocondriale richiede la sintesi di nucleotidi. L'attivazione genetica di Nrf2 migliora la biosintesi delle purine sovraregolando la via del pentoso fosfato e il metabolismo dei folati e della glutammina, in particolare nelle cellule in rapida proliferazione (Fig. 2) [24]. L'analisi del trascrittoma della drosofila mutante carente per la proteina chinasi mitocondriale serina / treonina chinasi putativa indotta da PTEN 1 (PINK1) ha dimostrato che la disfunzione mitocondriale porta alla sovraregolazione trascrizionale dei geni che influenzano il metabolismo dei nucleotidi [96], suggerendo che la biosintesi dei nucleotidi potenziata rappresenta un meccanismo di protezione contro le conseguenze neurotossiche della carenza di PINK1. Nrf2 regola l'espressione della fosforibosil pirofosfato amidotransferasi (PPAT), che catalizza l'ingresso nella via biosintetica dei nucleotidi purinici de novo e della metilenetetraidrofolato deidrogenasi 2 (MTHFD2) mitocondriale. Quest'ultimo è un enzima bifunzionale con attività deidrogenasi e cicloidrolasi che è fondamentale nel fornire sia glicina che formiato come fonti di unità a un carbonio per la biosintesi delle purine in cellule in rapida crescita [2]. È quindi probabile che l'attivazione di Nrf97 possa essere protettiva e potrebbe invertire la disfunzione mitocondriale nella carenza di PINK2. Infatti, l'attivazione farmacologica di Nrf1 da parte del sulforafano, o il triterpenoide RTA-2, ripristina e protegge le cellule carenti di PINK408 dalla tossicità della dopamina [1]. Sebbene i meccanismi sottostanti sembrino complessi, insieme, questi risultati indicano che l'attività Nrf98 può influenzare la biogenesi mitocondriale influenzando i livelli di espressione di fattori di trascrizione critici e coattivatori, nonché migliorando la biosintesi dei nucleotidi.
Nrf2 e integrità mitocondriale
Sebbene le prove dirette non siano sempre disponibili, vi sono forti indicazioni del fatto che Nrf2 è importante per l'integrità mitocondriale, in particolare in condizioni di stress ossidativo. I mitocondri isolati dal cervello e dal fegato di ratti a cui era stata somministrata una dose singola dell'attivatore Nrf2 sulforaphane sono resistenti all'apertura del poro di transizione della permeabilità mitocondriale (mPTP) causato dall'ossidante tert-butilidroperossido [99], [100]. L'mPTP, un complesso che consente alla membrana mitocondriale di diventare permeabile alle molecole con masse fino a 1500 Da, è stato recentemente identificato per essere formato da dimeri della F0F1-ATP sintasi [101]. La resistenza mediata dal sulforaphane all'apertura di mPTP è correlata all'aumento delle difese antiossidanti ei livelli di GSH mitocondriale, glutatione perossidasi 1, enzima malico 3 e tioredossina 2 sono tutti sovraregolati in frazioni mitocondriali isolate da animali trattati con sulforaphane [100].
Il danno alle proteine mitocondriali e il deterioramento della respirazione causati dal prodotto di perossidazione lipidica elettrofila 4-idrossi-2-nonenale sono attenuati nei mitocondri isolati dalla corteccia cerebrale dei topi trattati con sulforafano [102]. Nelle cellule epiteliali renali di ratto e nel rene, il sulforafano è protettivo contro la tossicità indotta da cisplatino e gentamicina e la perdita di µm [103], [104]. Durante il trattamento delle cellule muscolari lisce aortiche di ratto con sulforafano sono stati osservati anche protezione contro un pannello di ossidanti (superossido, perossido di idrogeno, perossinitrito) ed elettrofili (4-idrossi-2-nonenale e acroleina) e un aumento delle difese antiossidanti mitocondriali [105 ]. In un modello di danno renale acuto indotto da contrasto, è stato recentemente dimostrato che il precondizionamento ischemico degli arti ha effetti protettivi, inclusa l'inibizione dell'apertura dell'mPTP e il gonfiore mitocondriale, mediante l'attivazione di Nrf2 conseguente all'inibizione di GSK3? [106].
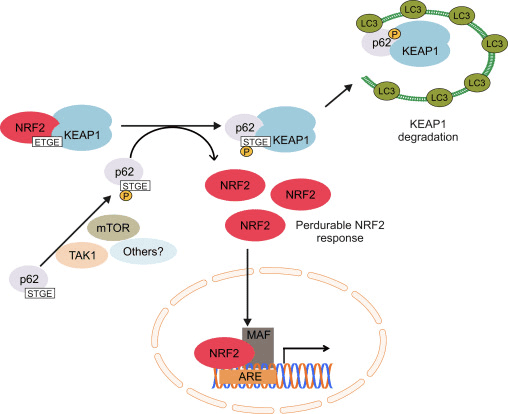
La mitofagia, il processo mediante il quale i mitocondri disfunzionali vengono selettivamente inghiottiti dagli autofagosomi e consegnati ai lisosomi per essere degradati e riciclati dalla cellula, è essenziale per l'omeostasi mitocondriale [107], [108]. Considerando che non è stata stabilita alcuna relazione causale tra Nrf2 e mitofagia, ci sono prove che il fattore di trascrizione può essere importante nel controllo della qualità mitocondriale giocando un ruolo nella mitofagia. Ciò potrebbe essere particolarmente evidente in condizioni di stress ossidativo. Pertanto, in un modello di sepsi, gli aumenti dei livelli della catena leggera 1-II del marker autofagosoma MAP3 (LC3-II) e della proteina di carico p62 a 24 ore dopo l'infezione sono soppressi in Nrf2-KO rispetto ai topi WT [109] . Recentemente è stato scoperto un induttore della mitofagia a piccole molecole (chiamato induttore della mitofagia mediato da p62, PMI); questo composto 1,4-difenil-1,2,3-triazolo è stato originariamente progettato come attivatore Nrf2 che interrompe l'interazione del fattore di trascrizione con Keap1 [110]. Simile alle cellule in cui Nrf2 è geneticamente sovraregolato (Keap1-KD o Keap1-KO), le cellule esposte a PMI hanno un riposo maggiore. È importante sottolineare che l'aumento della localizzazione mitocondriale LC3 che si osserva dopo il trattamento PMI delle cellule WT non si verifica nelle cellule Nrf2-KO, suggerendo il coinvolgimento di Nrf2.
Infine, l'analisi ultrastrutturale delle sezioni epatiche ha rivelato la presenza di mitocondri gonfiati con ridotta crista e membrane disgregate negli epatociti di Nrf2-KO, ma non in WT, topi che erano stati nutriti con una dieta ricca di grassi per le settimane 24; in particolare, questi fegati mostrano evidenti prove dello stress ossidativo e dell'infiammazione [68]. Si può concludere che Nrf2 ha un ruolo fondamentale nel mantenimento dell'integrità mitocondriale in condizioni di stress ossidativo e infiammatorio.
Sulforafano e suoi effetti su cancro, mortalità, invecchiamento, cervello e comportamento, malattie cardiache e altro
Gli isotiocianati sono alcuni dei composti vegetali più importanti che si possono ottenere nella dieta. In questo video faccio il caso più completo per loro che sia mai stato fatto. Soglia di attenzione breve? Passa al tuo argomento preferito facendo clic su uno dei seguenti punti temporali. Timeline completa di seguito.
Sezioni chiave:
00: 01: 14 - Cancro e mortalità
00: 19: 04 - Invecchiamento
00: 26: 30 - Cervello e comportamento
00: 38: 06 - Riassunto finale
00: 40: 27 - Dose
Timeline completa:
00: 00: 34 - Introduzione di sulforaphane, uno degli obiettivi principali del video.
00: 01: 14 - Consumo di verdure crocifere e riduzione della mortalità per tutte le cause.
00: 02: 12 - Rischio di cancro alla prostata.
00: 02: 23 - Rischio di cancro alla vescica.
00: 02: 34 - Carcinoma polmonare nei fumatori.
00: 02: 48 - Rischio di cancro al seno.
00: 03: 13 - Ipotetico: cosa succede se hai già un cancro? (Interventistica)
00: 03: 35 - Meccanismo plausibile che guida i dati associativi sul cancro e sulla mortalità.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Prova animale che mostra un forte effetto dell'estratto di germogli di broccolo sullo sviluppo del tumore della vescica nei ratti.
00: 06: 06 - Effetto dell'integrazione diretta di sulforafano nei pazienti affetti da cancro alla prostata.
00: 07: 09 - Bioaccumulo di metaboliti di isotiocianato nel tessuto mammario effettivo.
00: 08: 32 - Inibizione delle cellule staminali del carcinoma mammario.
00: 08: 53 - Lezione di storia: le brassiche sono state istituite come aventi proprietà sanitarie anche nell'antica Roma.
00: 09: 16 - La capacità del Sulforaphane di potenziare l'escrezione di cancerogeno (benzene, acroleina).
00: 09: 51 - NRF2 come interruttore genetico tramite elementi di risposta antiossidante.
00: 10: 10 - Come l'attivazione di NRF2 migliora l'escrezione di cancerogeno tramite glutatione-S-coniugati.
00: 10: 34 - I cavoletti di Bruxelles aumentano la glutatione-S-transferasi e riducono il danno al DNA.
00: 11: 20 - La bevanda di germogli di broccoli aumenta l'escrezione di benzene di 61%.
00: 13: 31 - L'omogenato di germogli di broccoli aumenta gli enzimi antiossidanti nelle vie aeree superiori.
00: 15: 45 - Consumo di verdure crocifere e mortalità per malattie cardiache.
00: 16: 55 - La polvere di germogli di broccoli migliora i lipidi nel sangue e il rischio complessivo di malattie cardiache nei diabetici di tipo 2.
00: 19: 04 - Inizio della sezione di invecchiamento.
00: 19: 21 - La dieta arricchita con sulforafano migliora la durata della vita dei coleotteri da 15 a 30% (in determinate condizioni).
00: 20: 34 - L'importanza della bassa infiammazione per la longevità.
00: 22: 05 - Le verdure crocifere e la polvere di germogli di broccoli sembrano ridurre un'ampia varietà di marcatori infiammatori negli esseri umani.
00: 23: 40 - Ricapitolazione di metà video: cancro, sezioni di invecchiamento
00: 24: 14 - Gli studi sui topi suggeriscono che il sulforafano potrebbe migliorare la funzione immunitaria adattativa in età avanzata.
00: 25: 18 - Sulforaphane ha migliorato la crescita dei peli in un modello murino di calvizie. Immagine su 00: 26: 10.
00: 26: 30 - Inizio della sezione cervello e comportamento.
00: 27: 18 - Effetto dell'estratto di germogli di broccoli sull'autismo.
00: 27: 48 - Effetto del glucorapanin sulla schizofrenia.
00: 28: 17 - Inizio della discussione sulla depressione (meccanismo e studi plausibili).
00: 31: 21 - Lo studio del mouse utilizzando 10 diversi modelli di depressione indotta da stress mostra sulforapano in modo simile efficace come fluoxetina (prozac).
00: 32: 00 - Lo studio mostra che l'ingestione diretta di glucorafanina nei topi è altrettanto efficace nel prevenire la depressione dal modello di stress sociale di sconfitta.
00: 33: 01 - Inizio della sezione di neurodegenerazione.
00: 33: 30 - Sulforaphane e malattia di Alzheimer.
00: 33: 44 - Sulforaphane e morbo di Parkinson.
00: 33: 51 - Sulforaphane e la malattia di Hungtington.
00: 34: 43 - Inizio della sezione traumatica di lesioni cerebrali.
00: 35: 01 - Sulforaphane iniettato immediatamente dopo TBI migliora la memoria (studio del mouse).
00: 35: 55 - Sulforaphane e plasticità neuronale.
00: 36: 32 - Sulforaphane migliora l'apprendimento nel modello di diabete di tipo II nei topi.
00: 37: 19 - Distrofia muscolare sulforapano e duchenne.
00: 37: 44 - Inibizione della miostatina nelle cellule muscolari satelliti (in vitro).
00: 38: 06 - Ricapitolazione tardiva: mortalità e cancro, danno al DNA, stress ossidativo e infiammazione, escrezione di benzene, malattie cardiovascolari, diabete di tipo II, effetti sul cervello (depressione, autismo, schizofrenia, neurodegenerazione), via NRF2.
00: 40: 27 - Pensieri sulla determinazione di una dose di germogli di broccoli o sulforafano.
00: 41: 01 - Aneddoti su germinazione a casa.
00: 43: 14 - Sulle temperature di cottura e sull'attività del sulforafano.
00: 43: 45 - Conversione batterica intestinale del sulforafano da glucorafanina.
00: 44: 24 - Gli integratori funzionano meglio se combinati con la mirosinasi attiva delle verdure.
00: 44: 56 - Tecniche di cottura e verdure crucifere.
00: 46: 06 - Isotiocianati come goitrogens.
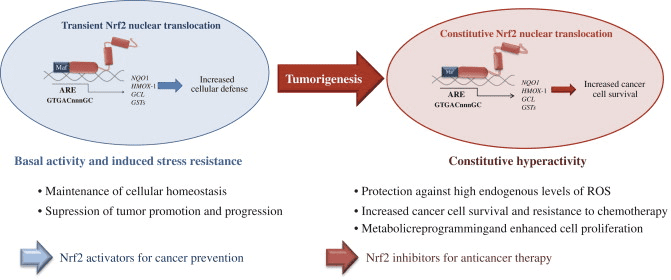
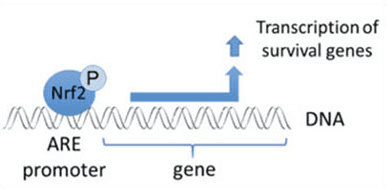
Nrf2 è un fattore di trascrizione che svolge un ruolo importante nel sistema di difesa antiossidante cellulare del corpo umano. L'elemento antiossidante reattivo, o ARE, è un meccanismo regolatore dei geni. Numerosi studi hanno dimostrato che Nrf2, o fattore 2 correlato a NF-E2, regola un'ampia varietà di geni basati su ARE in diversi tipi di cellule. È stato anche scoperto che Nrf2 svolge un ruolo essenziale nella protezione cellulare e nell'anti-cancerogenicità, dimostrando che Nrf2 può essere un trattamento efficace nella gestione delle malattie neurodegenerative e dei tumori che si ritiene siano causati dallo stress ossidativo. Dr. Alex Jimenez DC, CCST Insight
Osservazioni conclusive
Sebbene molte domande rimangano aperte, le prove sperimentali disponibili indicano chiaramente che Nrf2 è un attore importante nel mantenimento dell'omeostasi mitocondriale e dell'integrità strutturale. Questo ruolo diventa particolarmente critico in condizioni di stress ossidativo, elettrofilo e infiammatorio quando la capacità di sovraregolazione delle risposte citoprotettive mediate da Nrf2 influenza la salute generale e la sopravvivenza della cellula e dell'organismo. Il ruolo di Nrf2 nella funzione mitocondriale rappresenta un altro strato dei grandi meccanismi citoprotettivi orchestrati da questo fattore di trascrizione. Poiché molte condizioni patologiche umane hanno lo stress ossidativo, l'infiammazione e la disfunzione mitocondriale come componenti essenziali della loro patogenesi, l'attivazione farmacologica di Nrf2 è promettente per la prevenzione e il trattamento delle malattie. Comprensione completa dei meccanismi precisi con cui Nrf2 influenza la funzione mitocondriale è essenziale per la progettazione razionale di futuri studi clinici e può offrire nuovi biomarcatori per il monitoraggio dell'efficacia terapeutica.
Lo scopo dell'articolo sopra era quello di discutere, oltre a dimostrare, il ruolo emergente di Nrf2 nella funzione mitocondriale. Nrf2 o fattore nucleare fattore eritroide 2 correlato, è un regolatore emergente della resistenza cellulare agli ossidanti che possono contribuire allo stress ossidativo, influenzando la funzione cellulare e portando allo sviluppo di tossicità, malattie croniche e persino cancro. Mentre la produzione di ossidanti nel corpo umano può servire a vari scopi, tra cui la divisione cellulare, l'infiammazione, la funzione immunitaria, l'autofagia e la risposta allo stress, è essenziale controllare la loro sovrapproduzione per prevenire problemi di salute. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
Discussione aggiuntiva sull'argomento: Dolore alla schiena acuto
Mal di schienaè una delle cause più diffuse di disabilità e di giornate di lavoro perse in tutto il mondo. Il dolore alla schiena si attribuisce alla seconda ragione più comune per le visite mediche, superata solo dalle infezioni delle alte vie respiratorie. Circa l'80% della popolazione sperimenterà dolore alla schiena almeno una volta nella vita. La colonna vertebrale è una struttura complessa composta da ossa, articolazioni, legamenti e muscoli, tra gli altri tessuti molli. Per questo motivo, lesioni e / o condizioni aggravate, come dischi erniciati, può eventualmente portare a sintomi di mal di schiena. Gli infortuni sportivi o gli incidenti automobilistici sono spesso la causa più frequente di mal di schiena, tuttavia, a volte il più semplice dei movimenti può avere risultati dolorosi. Fortunatamente, opzioni di trattamento alternative, come la cura chiropratica, possono aiutare ad alleviare il mal di schiena attraverso l'uso di aggiustamenti spinali e manipolazioni manuali, migliorando in definitiva il sollievo dal dolore.
Nrf2 supporta l'attivazione di un gruppo di enzimi e geni antiossidanti e disintossicanti che proteggono il corpo umano dagli effetti di problemi di salute associati ad un aumento dei livelli di stress ossidativo, come il morbo di Alzheimer. È stata dimostrata una varietà di sostanze naturali per attivare il percorso Nrf2, che può aiutare a gestire i sintomi delle malattie neurodegenerative. Lo scopo di questo articolo è discutere il ruolo chiave di Nrf2 causato dall'infiammazione cronica.
Astratto
L'infiammazione è la caratteristica più comune di molte malattie e complicanze croniche, mentre svolge un ruolo critico nella cancerogenesi. Diversi studi hanno dimostrato che Nrf2 contribuisce al processo antinfiammatorio orchestrando il reclutamento di cellule infiammatorie e regolando l'espressione genica attraverso l'elemento di risposta antiossidante (ARE). La via di segnalazione Keap1 (proteina associata a ECH simile a Kelch) / Nrf2 (fattore 2 correlato a p45 NF-E2) / ARE regola principalmente l'espressione genica antinfiammatoria e inibisce la progressione dell'infiammazione. Pertanto, l'identificazione di nuovi fitochimici antinfiammatori dipendenti da Nrf2 è diventata un punto chiave nella scoperta di farmaci. In questa recensione, discutiamo i membri del percorso del segnale Keap1 / Nrf2 / ARE e dei suoi geni a valle, gli effetti di questo percorso su modelli animali di malattie infiammatorie e il crosstalk con il percorso NF-? B. Inoltre discutiamo anche della regolazione dell'inflammasoma NLRP3 da parte di Nrf2. Oltre a questo, riassumiamo lo scenario attuale dello sviluppo di sostanze fitochimiche antinfiammatorie e altri che mediano la via di segnalazione Nrf2 / ARE.
parole chiave: Nrf2, Keap1, ARE, Infiammazione, Stress ossidativo, Fitochimico
L'infiammazione è un processo complesso che si verifica quando i tessuti sono infettati o feriti da stimoli nocivi come agenti patogeni, danni o sostanze irritanti. Cellule immunitarie, vasi sanguigni e mediatori molecolari sono coinvolti in questa risposta protettiva [1]. L'infiammazione è anche un fenomeno patologico associato a una varietà di stati patologici indotti principalmente da fattori fisici, chimici, biologici e psicologici. Lo scopo dell'infiammazione è di limitare ed eliminare le cause del danno cellulare, cancellare e / o assorbire cellule e tessuti necrotici e iniziare la riparazione dei tessuti. Si distinguono due distinte forme di infiammazione: acuta e cronica. L'infiammazione acuta è auto-limitante e benefica per l'ospite, ma l'infiammazione cronica prolungata è una caratteristica comune di molte malattie croniche e complicanze. L'infiltrazione diretta di molte cellule immunitarie mononucleari come monociti, macrofagi, linfociti e plasmacellule, così come la produzione di citochine infiammatorie, porta a infiammazione cronica. È noto che l'infiammazione cronica svolge un ruolo fondamentale nella carcinogenesi [2]. In generale, sia i percorsi protesici che quelli antinfiammatori interagiscono nel normale processo infiammatorio.
Nel processo infiammatorio patologico vengono prima attivati mastociti, monociti, macrofagi, linfociti e altre cellule immunitarie. Quindi le cellule vengono reclutate nel sito della lesione, con conseguente generazione di specie reattive dell'ossigeno (ROS) che danneggiano le macromolecole compreso il DNA. Allo stesso tempo, queste cellule infiammatorie producono anche grandi quantità di mediatori infiammatori come citochine, chemochine e prostaglandine. Questi mediatori reclutano ulteriormente i macrofagi nei siti di infiammazione localizzati e attivano direttamente più cascate di trasduzione del segnale e fattori di trascrizione associati all'infiammazione. Le vie di segnalazione NF-? B (fattore nucleare kappa B), MAPK (proteina chinasi attivata da mitogeno) e JAK (janus chinasi) -STAT (trasduttori di segnale e attivatori di trascrizione) sono coinvolte nello sviluppo della via classica dell'infiammazione [3], [4], [5]. Studi precedenti hanno rivelato che il fattore di trascrizione Nrf2 (fattore 2 correlato a p45 NF-E2) regola l'espressione degli enzimi disintossicanti di fase II tra cui NADPH, NAD (P) H chinone ossidoreduttasi 1, glutatione perossidasi, ferritina, eme ossigenasi-1 (HO -1) e geni antiossidanti che proteggono le cellule da varie lesioni attraverso i loro effetti antinfiammatori, influenzando così il decorso della malattia [6], [7], [8].
Considerando questi notevoli risultati, lo sviluppo di farmaci terapeutici mirati per le malattie infiammatorie attraverso le vie di segnalazione ha suscitato molto interesse negli ultimi anni. In questa recensione, riassumiamo la ricerca sulla via di segnalazione di Keap1 (proteina associata a ECH associato a Kelch) / Nrf2 (fattore NF-E2 p45 correlato 2) / ARE (elemento di risposta antiossidante) nell'infiammazione.
Struttura e regolazione di Nrf2
Regolamento Nrf1 dipendente da Keap2
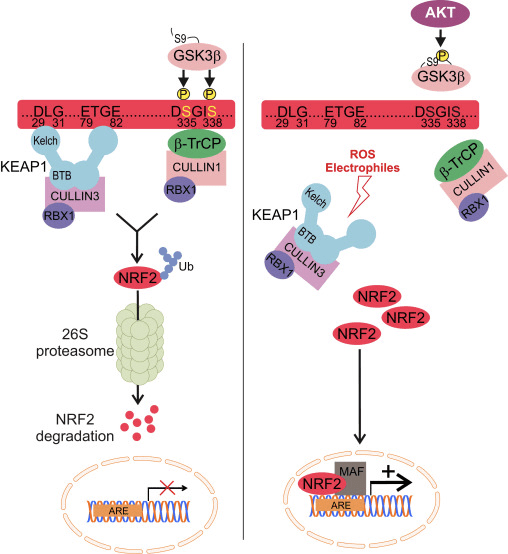
Nrf2 appartiene alla sottofamiglia Cap n Collar (CNC) e comprende sette domini funzionali, Neh (Omologia Nrf2-ECH) da 1 a Neh7 [9], [10]. Neh1 è un dominio CNC-bZIP che consente a Nrf2 di eterodimerizzare con piccole proteine musculoaponeurotiche fibrosarcoma (Maf), DNA e altri partner di trascrizione, oltre a formare un complesso nucleare con l'enzima ubiquitina-coniugante UbcM2 [11], [12]. Neh2 contiene due importanti motivi noti come DLG ed ETGE, che sono essenziali per l'interazione tra Nrf2 e il suo regolatore negativo Keap1 [13], [14].
Keap1 è un adattatore di substrato per l'ubiquitina ligasi E3 a base di cullina, che inibisce l'attività trascrizionale di Nrf2 tramite ubiquitinazione e degradazione proteasomale in condizioni normali [15], [16], [17]. I domini KELCH dell'omodimero Keap1 si legano con i motivi DLG ed ETGE del dominio Nrf2-Neh2 nel citosol, dove ETGE agisce come una cerniera con affinità più elevata e DLG agisce come un latch [18]. Sotto stress ossidativo o dopo esposizione a attivatori Nrf2, Nrf2 si dissocia dal legame Keap1 a causa della modifica tiolica dei residui di cisteina Keap1 che alla fine previene l'ubiquitinazione e la degradazione proteasomale di Nrf2 [19]. Quindi Nrf2 si traspone nel nucleo, eterodimerizza con piccole proteine Maf e transattiva una batteria di geni ARE (Fig. 1A). Il terminale carbossitico di Neh3 agisce come un dominio di transattivazione interagendo con il co-attivatore della trascrizione noto come CHD6 (proteina legante il DNA di cromo-ATPasi / elicasi) [20]. Neh4 e Neh5 agiscono anche come domini di transattivazione, ma si legano a un altro co-attivatore trascrizionale noto come CBP (proteina legante le proteine leganti la cAMP-risposta-elemento) [21]. Inoltre, Neh4 e Neh5 interagiscono con il cofattore nucleare RAC3 / AIB1 / SRC-3, portando a un'espressione genica ARE [2] potenziata da Nrf22. Neh5 ha un segnale di esportazione nucleare sensibile al redox che è cruciale per la regolazione e la localizzazione cellulare di Nrf2 [23].
Figura 1 Keap1-dipendente e regolazione indipendente di Nrf2. (A) In condizioni basali, Nrf2 viene sequestrato con Keap1 dai suoi due motivi (ETGE e DLG) che porta all'ubiquitinazione mediata da CUL3 seguita dalla degradazione del proteasoma. Sotto stress ossidativo, Nrf2 si dissocia da Keap1, si trasloca nel nucleo e attiva la batteria del gene ARE. (B) GSK3 fosforila Nrf2 e questo facilita il riconoscimento di Nrf2 da? -TrCP per l'ubiquitinazione mediata da CUL1 e la successiva degradazione del proteasoma. (C) p62 viene sequestrato con Keap1, portando alla sua degradazione autofagica, alla liberazione di Nrf2 e all'aumento della segnalazione Nrf2.
Regolamento Nrf1 indipendente da Keap2
Prove emergenti hanno rivelato un nuovo meccanismo di regolazione Nrf2 indipendente da Keap1. Il dominio Neh6 ricco di serina di Nrf2 gioca un ruolo cruciale in questa regolazione legandosi con i suoi due motivi (DSGIS e DSAPGS) alla proteina contenente ripetizione di? -Transducina (? -TrCP) [24]. ? -TrCP è un recettore substrato per il complesso ubiquitina ligasi Skp1 Cul1 Rbx1 / Roc1 che prende di mira Nrf2 per l'ubiquitinazione e la degradazione proteasomale. La glicogeno sintasi chinasi-3 è una proteina cruciale coinvolta nella stabilizzazione e regolazione di Nrf1 indipendente da Keap2; fosforila Nrf2 nel dominio Neh6 per facilitare il riconoscimento di Nrf2 da parte di? -TrCP e la successiva degradazione delle proteine [25] (Fig. 1B).
Altri regolatori Nrf2
Un'altra linea di evidenze ha rivelato un percorso non canonico di attivazione Nrf62 dipendente da p2 in cui p62 sequestra Keap1 a degradazione autofagica che alla fine porta alla stabilizzazione di Nrf2 e alla transattivazione dei geni dipendenti da Nrf2 [26], [27], [ 28], [29] (Fig. 1C).
Un numero sempre maggiore di prove suggerisce che diversi miRNA svolgono un ruolo importante nella regolazione dell'attività Nrf2 [30]. Sangokoya et al. [31] hanno dimostrato che il miR-144 sottoregola direttamente l'attività Nrf2 nella linea cellulare dei linfoblasti K562, nelle cellule progenitrici eritroidi umane primarie e nei reticolociti dell'anemia falciforme. Un altro interessante studio sulle cellule epiteliali del seno umano ha dimostrato che miR-28 inibisce Nrf2 attraverso un meccanismo indipendente da Keap1 [32]. Allo stesso modo, miRNA come miR-153, miR-27a, miR-142-5p e miR144 sottoregolano l'espressione di Nrf2 nella linea cellulare SH-SY5Y neuronale [33]. Singh et al. [34] hanno dimostrato che l'espressione ectopica di miR-93 diminuisce l'espressione dei geni regolati da Nrf2 in un modello di ratto di carcinogenesi mammaria indotto da 17? -Estradiolo (E2).
Una recente scoperta del nostro laboratorio ha identificato un inibitore endogeno di Nrf2 noto come recettore X retinoico alfa (RXR?). RXR? è un recettore nucleare, interagisce con il dominio Neh7 di Nrf2 (residui amminoacidici 209-316) tramite il suo dominio di legame al DNA (DBD) e inibisce specificamente l'attività Nrf2 nel nucleo. Inoltre, è stato riportato che altri recettori nucleari come il recettore attivato dal proliferatore del perossisoma, ER, recettore correlato agli estrogeni e i recettori glucocorticoidi sono inibitori endogeni dell'attività Nrf2 [9], [10].
Ruolo antinfiammatorio dell'asse Nrf2 / HO-1
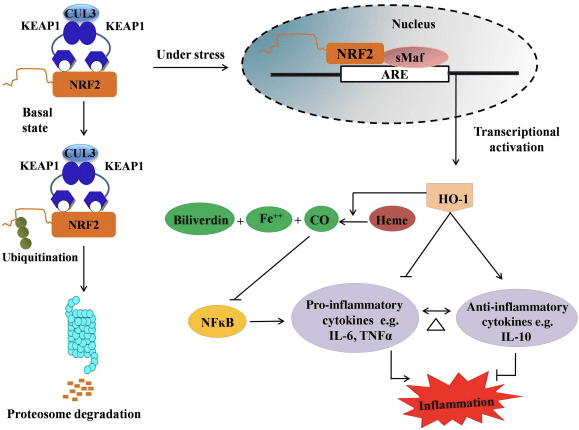
HO-1 è l'enzima inducibile e l'enzima limitante della velocità che catalizza la degradazione dell'eme in monossido di carbonio (CO) e ferro libero e biliverdina in bilirubina. Il degrado enzimatico dell'eme libera pro-infiammatoria e la produzione di composti anti-infiammatori come la CO e la bilirubina svolgono un ruolo importante nel mantenimento degli effetti protettivi di HO-1 (Fig. 2).
Figura 2 Panoramica del percorso Nrf2 / HO-1. In condizioni basali, Nrf2 si lega al suo repressore Keap1 che porta all'ubiquitinazione seguita dalla degradazione del proteasoma. Durante lo stress ossidativo, Nrf2 libero si trasloca nel nucleo, dove si dimerizza con i membri della piccola famiglia Maf e si lega ai geni ARE come HO-1. HO-1 sovraregolato catalizza l'eme in CO, bilirubina e ferro libero. Il CO agisce come un inibitore della via NF-? B che porta alla diminuzione dell'espressione delle citochine pro-infiammatorie, mentre la bilirubina agisce anche come antiossidante. Inoltre, HO-1 inibisce direttamente le citochine proinfiammatorie oltre ad attivare le citochine antinfiammatorie, portando così al bilanciamento del processo infiammatorio.
Nrf2 induce il gene HO-1 aumentando l'espressione di mRNA e proteine ed è uno dei classici geni regolati Nrf2 ampiamente utilizzato in numerosi studi in vitro e in vivo. Diversi studi hanno dimostrato che HO-1 e i suoi metaboliti hanno significativi effetti antinfiammatori mediati da Nrf2. L'aumento dell'espressione di HO-1, mediata da Nrf2 attivato, porta all'inibizione della segnalazione di NF-B, che si traduce in una ridotta lesione della mucosa intestinale e disfunzione della giunzione stretta nel modello di trapianto di fegato di ratto Sprague-Dawley maschile [35]. La sovraregolazione dell'espressione di HO-2 dipendente da Nrf1 può proteggere i mioblasti C2C12 derivati dal topo dalla citotossicità di H2O2 [36]. L'HO-2 dipendente da Nrf1 ha un impatto sulle risposte infiammatorie mediate dai lipopolisaccaridi (LPS) nei macrofagi cellulari schiumogeni RAW264.7 o derivati dai macrofagi peritoneali di topo. L'attività Nrf2 desensibilizza il fenotipo dei macrofagi delle cellule schiumose e previene l'infiammazione smodata dei macrofagi, che svolgono un ruolo importante nella progressione dell'aterosclerosi [37]. L'asse Nrf2 / HO-1 colpisce le cellule microgliali di topo BV2 indotte da LPS e le cellule HT22 dell'ippocampo di topo, con impatto sulla neuroinfiammazione. Sovraregolazione dell'espressione di HO-1 tramite la via Nrf2 nelle cellule microgliali BV2 di topo che difendono la morte cellulare delle cellule HT22 dell'ippocampo di topo [38]. Inoltre, le molecole ibride a base di cobalto (HYCO) che combinano un induttore Nrf2 con un rilascio di monossido di carbonio (CO) aumentano l'espressione di Nrf2 / HO-1, liberano CO ed esercitano attività antinfiammatoria in vitro. Gli HYCO regolano anche il tessuto HO-1 e rilasciano CO nel sangue dopo la somministrazione in vivo, supportando il loro potenziale utilizzo contro condizioni infiammatorie [39]. La sovraregolazione Nrf2 / HO-1 riduce l'infiammazione aumentando l'attività efferocitica dei macrofagi murini trattati con taurina clorammina [40]. Complessivamente, i modelli sperimentali sopra spiegati hanno rivelato che l'asse Nrf2 / HO-1 gioca un ruolo importante nella funzione antinfiammatoria, suggerendo che Nrf2 è un bersaglio terapeutico nelle malattie associate all'infiammazione.
Inoltre, i sottoprodotti di HO-1 come la CO, la bilirubina, agiscono come un potente antiossidante durante lo stress ossidativo e il danno cellulare [41], [42]; sopprime l'encefalomielite autoimmune e l'epatite [43], [44]; e protegge topi e ratti contro lo shock endotossico impedendo la generazione di iNOS e NO [45], [46], [47]. Inoltre, la bilirubina riduce l'attivazione e la disfunzione endoteliale [48]. È interessante notare che la bilirubina riduce la trasmigrazione dei leucociti endoteliali tramite la molecola di adesione 1 [49]. Questi riferimenti specifici che indicano non solo HO-1 agisce come un potente agente antinfiammatorio ma anche i suoi metaboliti.
Mediatori infiammatori ed enzimi inibiti da Nrf2
Citochine e chemochine
Le citochine sono proteine a basso peso molecolare e polipeptidi secreti da una varietà di cellule; regolano la crescita cellulare, la differenziazione e la funzione immunitaria e sono coinvolti nell'infiammazione e nella guarigione delle ferite. Le citochine includono interleuchine (IL), interferoni, fattore di necrosi tumorale (TNF), fattore stimolante le colonie, chemochine e fattori di crescita. Alcune citochine sono considerate mediatori pro-infiammatori mentre altre hanno funzioni antinfiammatorie. L'esposizione allo stress ossidativo provoca la sovrapproduzione di citochine che causa stress ossidativo nelle cellule bersaglio. Diverse citochine pro-infiammatorie vengono prodotte in eccesso quando NF-? B viene attivato dallo stress ossidativo. Inoltre, lo stress ossidativo pro-infiammatorio provoca un'ulteriore attivazione di NF-? B e la sovrapproduzione di citochine. L'attivazione del sistema Nrf2 / ARE gioca un ruolo importante nell'interrompere questo ciclo. Le chemochine sono una famiglia di piccole citochine, il cui ruolo principale è guidare la migrazione delle cellule infiammatorie. Funzionano principalmente come agenti chemiotattici per leucociti, monociti, neutrofili e altre cellule effettrici.
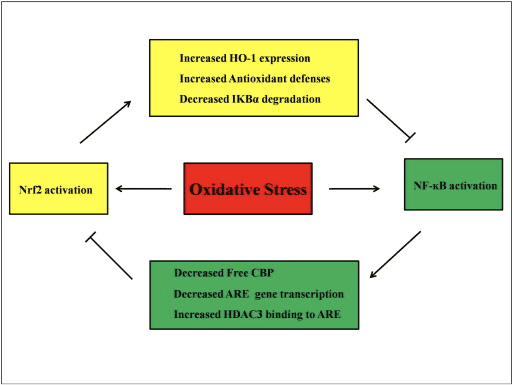
È stato riportato che l'attivazione di Nrf2 impedisce la sovraregolazione trascrizionale indotta da LPS delle citochine pro-infiammatorie, comprese IL-6 e IL-1? [50]. IL-1? e la produzione di IL-6 è aumentata anche in Nrf2? /? topi con colite indotta da destrano solfato [51], [52]. Nrf2 inibisce la produzione di IL-17 a valle e altri fattori infiammatori Th1 e Th17 e sopprime il processo patologico in un modello sperimentale di sclerosi multipla, l'encefalite autoimmune [53]. I geni antiossidanti Nrf2-dipendenti HO-1, NQO-1, Gclc e Gclm bloccano TNF-?, IL-6, la proteina 1 attrattiva della chemio monocita (MCP1), la proteina infiammatoria 2 dei macrofagi (MIP2) e l'infiammazione mediatori. Ma nel caso dei topi knockout Nrf2, l'effetto antinfiammatorio non si verifica [54]. I neutrofili peritoneali da topi knockout Nrf2 trattati con LPS hanno livelli significativamente più alti di citochine (TNF-? E IL-6) e chemochine (MCP1 e MIP2) rispetto alle cellule wild-type (WT) [54]. In vitro, il trasferimento del gene Nrf2 a cellule muscolari lisce aortiche umane e di coniglio sopprime la secrezione di MCP1 [8], [55] e l'espressione di HO-2 dipendente da Nrf1 sopprime TNF -? - stimolato NF-? B e MCP-1 secrezione nelle cellule endoteliali della vena ombelicale umana [56]. Questi risultati suggeriscono che, in risposta a stimoli infiammatori, la sovraregolazione della segnalazione Nrf2 inibisce la sovrapproduzione di citochine e chemochine pro-infiammatorie, oltre a limitare l'attivazione di NF-? B.
Molecole di adesione cellulare
Le molecole di adesione cellulare (CAM) sono proteine che si legano alle cellule o alla matrice extracellulare. Situati sulla superficie cellulare, sono coinvolti nel riconoscimento cellulare, nell'attivazione cellulare, nella trasduzione del segnale, nella proliferazione e nella differenziazione. Tra i CAM, ICAM-1 e VCAM-1 sono membri importanti della superfamiglia delle immunoglobuline. L'ICAM-1 è presente in basse concentrazioni nelle membrane delle cellule leucocitarie e endoteliali. Dopo la stimolazione delle citochine, la concentrazione aumenta in modo significativo. L'ICAM-1 può essere indotto da IL-1 e TNF ed è espresso dall'endotelio vascolare, dai macrofagi e dai linfociti. È un ligando dell'integrina, un recettore presente sui leucociti. Quando il ponte ICAM-1-integrina viene attivato, i leucociti si legano alle cellule endoteliali e quindi migrano nei tessuti subendoteliali [57]. VCAM-1 media l'adesione di linfociti, monociti, eosinofili e basofili all'endotelio vascolare e contribuisce al reclutamento dei leucociti, che alla fine porta a danni ai tessuti dovuti allo stress ossidativo. Nrf2 inibisce l'attività del promotore di VCAM-1 [58]. Il gene a valle HO-2 regolato da Nrf1 può influenzare l'espressione di E-selectina e VCAM-1, molecole di adesione associate alle cellule endoteliali [59]. L'espressione polmonare di diverse CAM come CD-14, TREM1, SELE, SELP e VCAM-1 sono significativamente più alte in Nrf2? /? topi rispetto ai topi Nrf2 + / + [60]. Nrf2 nelle cellule endoteliali aortiche umane sopprime l'espressione di VCAM-1 indotta da TNF -? - e interferisce con l'adesione delle cellule monocitiche U937 indotte da TNF -? La sovraespressione di Nrf8 inibisce anche l'espressione genica VCAM-2 indotta da TNF -? - nelle cellule endoteliali microvascolari umane [1]. Si trova che l'acido 61-idrossiantranilico (HA) antiossidante naturale, uno dei metaboliti dell'l-triptofano formati in vivo lungo la via metabolica nota come via della chinurenina durante l'infiammazione o l'infezione, induce l'espressione di HO-3 e stimola Nrf1 nell'ombelicale umano cellule endoteliali della vena (HUVEC). L'espressione di HO-2 dipendente da Nrf2 indotta da HA inibisce la secrezione di MCP-1, l'espressione di VCAM-1 e l'attivazione di NF-kB associata a danno vascolare e infiammazione nell'aterosclerosi [1]. Il derivato calcone sintetico antiproliferativo e antinfiammatorio 56?, 2?, 4? -Tris (metossimetossi) calcone inibisce ICAM-6, la citochina proinfiammatoria IL-1? E TNF-? espressione nel tessuto del colon di topi trattati con acido trinitrobenzensolfonico [1]. La sovraregolazione di Nrf62 inibisce l'espressione di ICAM-2 indotta da TNF -? - nelle cellule epiteliali pigmentate retiniche umane trattate con licopene [1]. Tutti questi studi suggeriscono che Nrf63 svolge un ruolo chiave nel processo infiammatorio regolando la migrazione e l'infiltrazione delle cellule infiammatorie nel tessuto infiammato.
Matrix Metalloproteinases (MMP)
Le MMP sono ampiamente presenti nella matrice extracellulare e sono coinvolte in processi fisiologici e patologici come la proliferazione cellulare, la migrazione, il differenziamento, la guarigione delle ferite, l'angiogenesi, l'apoptosi e le metastasi tumorali. È stato riportato che l'asse Nrf2 / HO-1 inibisce MMP-9 nei macrofagi e MMP-7 nelle cellule epiteliali intestinali umane, e questo è utile nel trattamento della malattia infiammatoria intestinale [62], [64]. Il danno cutaneo indotto dall'irradiazione UV è più grave nei topi knockout Nrf2 rispetto ai topi WT e il livello di MMP-9 è significativamente più alto, indicando che Nrf2 riduce l'espressione di MMP-9. Pertanto, Nrf2 è considerato protettivo contro l'irradiazione UV [65]. Un altro studio ha anche riportato che l'attivazione trascrizionale sottoregolata di MMP-9 nell'invasione e nell'infiammazione delle cellule tumorali è regolata attraverso l'inibizione della via di segnalazione NF-kB [66]. Nella lesione traumatica del midollo spinale, la via di segnalazione NF-kB partecipa anche alla regolazione dei livelli di mRNA di MMP-9 [67]. Pertanto, nell'infiammazione la regolazione delle MMP è influenzata direttamente dalla via Nrf2 o indirettamente attraverso la via NF-? B influenzata da Nrf2.
Cyclooxygenase-2 (COX2) e Inducible Nitric Oxide Synthase (INOS)
Una serie di esperimenti su topi knockout Nrf2 hanno dimostrato il suo ruolo cruciale nell'infiammazione e nella regolazione di geni pro-infiammatori come COX-2 e iNOS. Per la prima volta, Khor et al. riportato una maggiore espressione di citochine pro-infiammatorie come COX-2 e iNOS nei tessuti del colon di Nrf2? /? topi rispetto ai topi WT Nrf2 + / +, indicando che Nrf2 sopprime la loro attività [51]. Un altro rapporto sul pretrattamento con sulforafano, uno dei ben noti attivatori Nrf2 presenti nelle verdure crocifere, ha dimostrato il suo effetto antinfiammatorio di inibire l'espressione di TNF-?, IL-1 ?, COX-2 e iNOS a entrambi gli mRNA e livelli di proteine nei macrofagi peritoneali primari da topi Nrf2 + / + rispetto a quelli da Nrf2? /? topi [68]. Allo stesso modo, l'ippocampo di topi knockout Nrf2 con infiammazione indotta da LPS mostra anche una maggiore espressione di marcatori di infiammazione come iNOS, IL-6 e TNF-? rispetto ai topi WT [69]. Allo stesso modo, i topi knockout Nrf2 sono ipersensibili allo stress ossidativo indotto dalla 1-metil-4-fenil-1,2,3,6-tetraidropiridina e mostrano un aumento dei livelli di mRNA e proteine dei marcatori di infiammazione come COX-2, iNOS , IL-6 e TNF-? [70]. Inoltre, fegati di Nrf2? /? i topi stimolati con una dieta carente di metionina e colina hanno un'espressione di mRNA di Cox5 e iNOS ~ 2 volte più alta rispetto a quelli dei topi WT con la stessa dieta, suggerendo un ruolo antinfiammatorio di Nrf2 [71]. Recentemente, Kim et al. ha dimostrato che l'etil piruvato fitochimico esercita i suoi effetti antinfiammatori e antiossidanti diminuendo l'espressione di iNOS attraverso la segnalazione Nrf2 nelle cellule BV2. Hanno dimostrato che l'etil piruvato induce la traslocazione nucleare di Nrf2, che alla fine inibisce l'interazione tra p65 e p300, portando a una diminuzione dell'espressione di iNOS [72]. Inoltre, l'analogo del carbazolo LCY-2-CHO attiva Nrf2 e causa la sua traslocazione nucleare, portando alla soppressione dell'espressione di COX2 e iNOS [73] nelle cellule muscolari lisce vascolari aortiche di ratto.
Ruolo paradossale di Nrf2 nel regolamento di NLRP3 iIflammasome Activity
La famiglia NLR, dominio pirinico contenente 3 (NLRP3) inflammasoma è un complesso multiproteico che funziona come un recettore di riconoscimento dei patogeni (PRR) e riconosce l'ampia gamma di segnali di stress ossidativo microbico come i pattern molecolari associati ai patogeni (PAMP), Danni- molecole di pattern molecolari associate (DAMP) e ROS [74]. L'inflammasoma NLRP3 attivato media la scissione della caspasi-1 e la secrezione della citochina pro-infiammatoria interleuchina-1? (IL-1?) Che alla fine induce il processo di morte cellulare noto come piroptosi che protegge gli ospiti da un'ampia gamma di agenti patogeni [75]. Tuttavia, l'attivazione aberrante dell'inflammasoma è associata a malattie da ripiegamento errato delle proteine come encefalopatie spongiformi trasmissibili, morbo di Alzheimer, morbo di Parkinson e anche diabete di tipo 2 [76], cancro [77], gotta e aterosclerosi [78].
Una recente osservazione del gruppo Rong Hu sull'associazione di Nrf2 con la regolazione negativa dell'inflammasoma ha rivelato che Nrf2 induce l'espressione di NQO1 che porta all'inibizione dell'attivazione dell'inflammasoma NLRP3, della scissione della caspasi-1 e dell'IL-1? generazione nei macrofagi. Inoltre, un ben noto attivatore Nrf2, tert-butilidrochinone (tBHQ) ha regolato negativamente la trascrizione NLRP3 attivando l'ARE in modo Nrf2-dipendente [79]. Oltre all'osservazione di cui sopra, lo stesso gruppo è stato anche rivelato che il dimetilfumarato (DMF) previene la colite indotta da DSS attivando la via di segnalazione Nrf2 che è coinvolta nella traslocazione nucleare Nrf2 e nell'inibizione dell'assemblaggio dell'inflammasoma NLRP3 [80].
Una serie di esperimenti che utilizzano composti naturali e sintetici hanno anche rivelato l'effetto inibitorio di Nrf2 sull'attivazione dell'inflammasoma NLRP3. Ad esempio, il trattamento dell'epigallocatechina-3-gallato (EGCG) nei topi con nefrite lupica ha dimostrato di diminuire l'attivazione dell'inflammasoma NLRP3 renale che è mediata dalla via di segnalazione Nrf2 [81]. Allo stesso modo, il citrale (3,7-dimetil-2,6-ottadienale), un importante composto attivo in una medicina erboristica cinese Litsea cubeba, inibisce l'attivazione dell'inflammasoma NLRP3 tramite la via di segnalazione antiossidante Nrf2 nel modello murino accelerato e grave di nefrite lupica (ASLN) [82]. Allo stesso modo, la biocanina ha protetto contro il danno epatico indotto da LPS / GalN attivando la via Nrf2 e inibendo l'attivazione dell'inflammasoma NLRP3 nei topi maschi BALB / c [83]. Inoltre, è stato anche dimostrato che la mangiferina sovra-regola l'espressione di Nrf2 e HO-1 in modo dose-dipendente e ha inibito NLRP3 epatico indotto da LPS / D-GalN, ASC, caspase-1, IL-1? e TNF-? espressione [84].
Nonostante la regolazione negativa di NLRP3 da parte di Nrf2, attiva anche la funzione inflammasoma NLRP3 e AIM2. Haitao Wen e colleghi lo hanno scoperto, Nrf2? /? i macrofagi di topo hanno mostrato l'attivazione difettosa dell'inflammasoma NLRP3 e AIM2 ma non dell'inflammasoma NLRC4 [85]. È interessante notare che questa osservazione descrive le funzioni sconosciute di Nrf2 nel contesto delle malattie associate all'infiammazione; quindi è molto importante studiare ulteriormente per rivelare il meccanismo in cui Nrf2 attiva la funzione inflammasoma prima di considerarlo come un target terapeutico.
Soppressione della trascrizione di citochine proinfiammatorie da parte di Nrf2
Un'indagine molto recente basata sui risultati dell'immunoprecipitazione della cromatina (ChIP) -seq e ChIP-qPCR nei macrofagi del topo ha rivelato che Nrf2 si lega alle regioni promotrici di citochine pro-infiammatorie come IL-6 e IL-1? e inibisce il reclutamento di RNA Pol II. Di conseguenza, RNA Pol II non è in grado di elaborare l'attivazione trascrizionale di IL-6 e IL-1? che alla fine porta all'inibizione dell'espressione genica. Per la prima volta, il gruppo di Masayuki Yamamoto ha rivelato il nuovo meccanismo mediante il quale Nrf2 non solo transattiva i suoi geni a valle attraverso ARE, ma sopprime anche l'attivazione trascrizionale di geni specifici con o senza ARE attraverso l'inibizione del reclutamento di RNA Pol II [50].
Diafonia tra Nrf2 e NF-? B Pathways
NF-? B è un complesso proteico responsabile della trascrizione del DNA che si trova in quasi tutti i tipi di cellule animali e coinvolto in vari processi come infiammazione, apoptosi, risposta immunitaria, crescita cellulare e sviluppo. p65, una proteina Rel della famiglia NF-? B, ha un dominio di transattivazione mentre p50 no e richiede l'eterodimerizzazione con la proteina Rel per attivare la trascrizione. Durante lo stress ossidativo, la I? B chinasi (IKK) viene attivata e provoca la fosforilazione di I? B, con conseguente rilascio e traslocazione nucleare di NF-? B. NF-? B provoca la trascrizione di mediatori pro-infiammatori come IL-6, TNF-?, INOS, IL-1 e adesione intracellulare COX-2.
La regolazione anormale di NF-? B è stata collegata ad artrite reumatoide, asma, malattie infiammatorie intestinali e gastrite indotta da infezione da Helicobacter pylori [86]. Si ritiene attualmente che l'attività di NF-kB influenzi la via di segnalazione Keapl / Nrf2 / ARE principalmente in tre aspetti: in primo luogo, Keap1 degrada IKK? attraverso l'ubiquitinazione, inibendo così l'attività di NF-? B [87]. In secondo luogo, il processo infiammatorio induce mediatori infiammatori come COX2 derivato dal ciclopentenone prostaglandina 15d-PGJ2, un forte elettrofilo che reagisce con Keap1 e attiva Nrf2, avviando così la trascrizione genica con simultanea inibizione dell'attività NF-kB [58], [88] ( Fig.3 A, B). Terzo, NF-? B può combinarsi con il coattivatore trascrizionale competitivo Nrf2 CBP [89], [90] (Fig. 3 C, D).
Figura 3 Diafonia tra i percorsi Nrf2 e NF-? B. (A) Keap1 dirige l'IKK verso l'ubiquitinazione mediata da CUL3 e la degradazione del proteasoma che alla fine porta all'inibizione della fosforilazione di NF-? B e questo meccanismo funziona anche come legame competitivo di Nrf2 e IKK con Keap1. (B) Lo stress ossidativo attiva IKK che fosforila NF-? B, portando alla sua traslocazione nel nucleo e all'attivazione di citochine proinfiammatorie come COX-2. Il prodotto terminale della COX-2 noto come 15d-PGJ2 agisce come un induttore di Nrf2 che alla fine porta alla soppressione dello stress ossidativo. (C) Nrf2 si lega al suo cofattore trascrizionale CBP insieme al piccolo Maf e ad altri macchinari trascrizionali per avviare l'espressione genica guidata da ARE. (D) Quando NF-? B si lega con CBP in modo competitivo, inibisce il legame di CBP con Nrf2, che porta all'inibizione della transattivazione di Nrf2.
Si presume che le vie di segnalazione Nrf2 e NF-? B interagiscano per controllare la trascrizione o la funzione delle proteine bersaglio a valle. A giustificazione di questa ipotesi, molti esempi mostrano che l'attivazione e l'inibizione diretta o indiretta si verificano tra i membri delle vie Nrf2 e NF-? B (Fig. 4). In risposta a LPS, il knockdown di Nrf2 aumenta significativamente l'attività trascrizionale di NF-? B e la trascrizione del gene dipendente da NF-? B, dimostrando che Nrf2 impedisce l'attività di NF-? B [60], [91]. Inoltre, una maggiore espressione di HO-2 a valle dipendente da Nrf1 inibisce l'attività NF-? B. Quando le cellule del cancro alla prostata sono brevemente esposte a? -Tochoferil succinato, un derivato della vitamina E, l'espressione di HO-1 è sovraregolata. I prodotti finali di HO-1 inibiscono la traslocazione nucleare di NF-? B [92]. Questi studi in vivo suggeriscono che Nrf2 regola negativamente la via di segnalazione NF-kB. LPS stimola l'attività di legame del DNA NF-? B e il livello della subunità p65 di NF-? B è significativamente più alto negli estratti nucleari dai polmoni di Nrf2? /? rispetto ai topi WT, suggerendo un ruolo negativo di Nrf2 nell'attivazione di NF-? B. Inoltre, Nrf2? /? fibroblasti embrionali di topo trattati con LPS e TNF-? mostrano più prominente attivazione NF-? B causata dall'attivazione IKK e I? B-? degradazione [60]. E la clearance del virus respiratorio sinciziale è significativamente ridotta mentre l'attività di legame del DNA di NF-? B è aumentata in Nrf2? /? topi rispetto ai topi WT [93]. Nefrite lupica indotta da Pristane in Nrf2? /? i topi co-trattati con sulforafano hanno gravi danni renali e alterazioni patologiche, nonché un'elevata espressione di iNOS e attivazione di NF-? B rispetto al WT, suggerendo che Nrf2 migliora la nefrite lupica inibendo la via di segnalazione NF-? B e eliminando ROS [94 ]. L'attività di NF-? B si verifica anche quando le cellule vengono trattate con un induttore Nrf2 insieme a LPS e TNF- ?. Ad esempio, un derivato sintetico del calcone inibisce l'attivazione di NF-? B indotta dal TNF-α sia direttamente che indirettamente e in parte attraverso l'induzione dell'espressione di HO-1 nelle cellule HT-29 epiteliali intestinali umane [62]. La soppressione della traslocazione di NF-? B e dell'attività di legame al DNA così come la soppressione dell'espressione di iNOS negli epatociti si trovano quando i ratti F344 vengono trattati con 3H-1,2-ditiolo-3-tione (D3T) [95]. Dopo il co-trattamento con sulforafano e LPS, l'espressione indotta da LPS di iNOS, COX-2 e TNF-? in Raw 264.7 i macrofagi sono sottoregolati, suggerendo che il sulforafano ha attività antinfiammatoria attraverso l'inibizione del legame del DNA NF-? B [96]. Sebbene siano stati condotti diversi studi sperimentali per spiegare il collegamento tra i percorsi Nrf2 e NF-? B, rimangono risultati contrastanti. Sono state riportate entrambe le norme positive e negative tra Nrf2 e NF-kB [97]. Di solito, gli elettrofili chemiopreventivi 3H-1,2-dithiole-3-thione, sulforaphane e Triterpenoid CDDO-Me attivano Nrf2 inibendo NF-kB e i suoi geni downregulated [98], [99], [100]. Al contrario, è stato dimostrato che diversi agenti o condizioni come ROS, LPS, stress di taglio del flusso, LDL ossidato e fumo di sigaretta aumentano sia l'attività di Nrf2 che quella di NF-kB [97]. Inoltre, studi in vivo hanno rivelato che l'attività di NF-kB è ridotta nei fegati isolati da Nrf2? /? topi e l'attività di legame NF-? B è inferiore in Nrf2? /? rispetto ai topi Nrf2 + / + [101]. Tuttavia, le cellule endoteliali aortiche umane trattate con il vettore adenovirale Nrf2 inibiscono i geni a valle di NF-? B senza influenzare l'attività di NF-? B [8].
Figura 4 Circuito normativo di Nrf2 e NF-? B. La via Nrf2 inibisce l'attivazione di NF-? B prevenendo la degradazione di I? B-? e aumentando l'espressione di HO-1 e le difese antiossidanti che neutralizzano i ROS e le sostanze chimiche disintossicanti. Di conseguenza, l'attivazione di NF-? B associata a ROS viene soppressa. Allo stesso modo, la trascrizione mediata da NF-? B riduce l'attivazione di Nrf2 riducendo SONOTrascrizione genica e proteina legante CREB libera competendo con Nrf2 per CBP. Inoltre, NF-? B aumenta il reclutamento dell'istone deacetilasi (HDAC3) nella regione ARE e quindi viene impedita l'attivazione trascrizionale di Nrf2.
L'attivazione della via di segnalazione Nrf2 gioca un ruolo importante nell'espressione di enzimi e geni coinvolti nella detossificazione degli ossidanti reattivi, migliorando la capacità antiossidante delle cellule nel corpo umano. Mentre molti studi di ricerca sono oggi disponibili, i meccanismi regolatori nell'attivazione di Nrf2 non sono completamente compresi. È stato anche scoperto un possibile ruolo della via di segnalazione Nrf2 nel trattamento dell'infiammazione. Dr. Alex Jimenez DC, CCST Insight
Ruolo di Nrf2 nelle malattie infiammatorie
Studi in vivo hanno dimostrato che Nrf2 svolge un ruolo importante nelle malattie infiammatorie che colpiscono diversi sistemi; questi includono gastrite, colite, artrite, polmonite, danni al fegato, malattie cardiovascolari, malattie neurodegenerative e danni al cervello. In questi studi, Nrf2? /? gli animali hanno mostrato sintomi più gravi di infiammazione e danni ai tessuti rispetto agli animali WT. Pertanto, si ritiene che la via di segnalazione Nrf2 abbia un effetto protettivo nelle malattie infiammatorie. L'installazione intra-tracheale dell'elastasi porcina pancreatica induce patologia polmonare ostruttiva cronica, in particolare enfisema. I topi con deficit di Nrf2 sono altamente suscettibili all'enfisema e la ridotta espressione di HO-1, PrxI e il gene SLPI antiprotease si verificano nei macrofagi alveolari. Nrf2 è considerato un regolatore chiave nel sistema di difesa mediato dai macrofagi contro il danno polmonare [102]. Topi affetti da Nrf2 con enfisema indotto dall'esposizione al fumo di tabacco per 6 mesi mostrano una maggiore infiammazione broncoalveolare, espressione sovraregolata di marcatori di stress ossidativo negli alveoli e aumento dell'apoptosi delle cellule del setto alveolare, suggerendo che Nrf2 agisce contro l'enfisema indotto da tabacco attraverso l'aumentata espressione di antiossidante geni [102], [103]. Con l'interruzione di Nrf2, l'infiammazione delle vie aeree allergica e l'asma che utilizza il complesso ovaluminoso mostrano un'aumentata infiammazione delle vie aeree, iper-reattività delle vie aeree, iperplasia delle cellule caliciformi e alti livelli di Th2 nel lavaggio broncoalveolare e splenociti, mentre la via di segnalazione mediata da Nrf2 limita l'eosinofilia delle vie aeree , ipersecrezione del muco e iper-reattività delle vie aeree oltre a indurre molti geni antiossidanti che impediscono lo sviluppo di asma [104]. L'iniezione di carragenina nella cavità pleurica induce la pleurite e l'accumulo di 15d-PGJ2 nelle cellule infiammatorie Nrf2 è limitato ai macrofagi peritoneali di topo. Durante la fase iniziale dell'infiammazione, 15d-PGJ2 attiva Nrf2 e regola il processo infiammatorio attraverso l'induzione di HO-1 e PrxI. Uno studio ha anche suggerito che COX-2 ha un effetto anti-infiammatorio nella fase iniziale dalla produzione di 15d-PGJ2 [105]. La somministrazione orale di 1% destrano solfato di sodio per 1 settimana induce la colite associata a alterazioni istologiche che includono l'accorciamento delle cripte e l'infiltrazione delle cellule infiammatorie nel tessuto del colon. Per proteggere l'integrità intestinale nella colite, Nrf2 potrebbe svolgere un ruolo importante regolando le citochine pro-infiammatorie e inducendo enzimi disintossicanti di fase II [51]. In un modello murino knockout Nrf2 di sepsi polmonare indotta da LPS, l'attività NF-? B regola l'influenza delle citochine infiammatorie come COX-2, IL-113, IL-6 e TNF? che sono essenziali per iniziare e promuovere l'infiammazione [60]. Nrf2 riduce il danno infiammatorio regolando questi fattori infiammatori. In questi modelli di infiammazione acuta, l'aumento della regolazione degli enzimi antiossidanti, delle citochine pro-infiammatorie e dei mediatori attraverso la via di segnalazione Nrf2 riduce la lesione infiammatoria negli animali WT. È interessante notare che questo è stato anche riportato in topi knockout Nrf2 in cui i sintomi sono marcatamente esacerbati rispetto ai topi WT.
Ricerca sui farmaci antinfiammatori dipendenti da Nrf2
In sintesi, abbiamo discusso di esperimenti che dimostrano che la via del segnale Nrf2 svolge un ruolo regolatore in molte aree di infiammazione, quindi gli agenti antinfiammatori Nrf2-dipendenti sono importanti per il trattamento delle malattie infiammatorie.
Le piante sono state straordinariamente ricche fonti di composti che attivano il fattore di trascrizione Nrf2, portando all'up-regulation dei geni citoprotettivi. Recentemente, sono stati condotti diversi studi per studiare gli effetti di diversi agenti anti-infiammatori, per lo più di origine vegetale. Ad esempio, la curcumina è l'ingrediente attivo della curcuma e si trova anche in piccole quantità nello zenzero; isotiocianati, in particolare fenilisotiocianati sono da broccoli, sedano e altre verdure; e gli antociani provengono da bacche e uva [124]. Gli studi hanno dimostrato che tutti questi agenti non sono solo buoni antiossidanti ma hanno anche potenti effetti anti-infiammatori attraverso l'induzione di Nrf2 [125], [126]. Pertanto, lo sviluppo di nuovi attivatori antinfiammatori Nrf2 dall'estratto vegetale ha suscitato molto interesse nella ricerca medica.
Negli ultimi anni sono stati condotti molti esperimenti sugli animali per confermare le azioni di questi composti. Artesunate è utilizzato principalmente per la malaria grave, la malaria cerebrale e le malattie autoimmuni reumatiche; è anche efficace nel danno polmonare settico. L'artesunato attiva l'espressione di Nrf2 e HO-1 e quest'ultima riduce l'afflusso di citochine pro-infiammatorie e leucociti nei tessuti per prevenire l'infiammazione [127]. Si ritiene che l'isovitexina, estratta dalle carene del riso Oryza sativa, abbia proprietà antinfiammatorie e antiossidanti; svolge un ruolo protettivo contro il danno polmonare acuto indotto da LPS attivando la via Nrf2 / HO-1 e inibendo MAPK e NF-? B [128]. Fimasartan, un recentemente popolare bloccante del recettore dell'angiotensina II che agisce sul sistema renina-angiotensina, riduce la pressione sanguigna; l'utilizzo di fimasartan per il trattamento di topi con ostruzione ureterale unilaterale indotta chirurgicamente riduce lo stress ossidativo, l'infiammazione e la fibrosi attraverso la sovraregolazione di Nrf2 e la via antiossidante e l'inibizione di RAS e MAPK [129]. Sappanone è ampiamente distribuito nel sud-est asiatico, dove viene utilizzato come farmaco anti-influenzale, antiallergico e neuroprotettivo; attiva Nrf2 e inibisce NF-? B e quindi può essere utile nel trattamento delle malattie correlate a Nrf2- e / o NF-? B [130]. La bissina estratta dai semi di Bixin orellana è utilizzata per malattie infettive e infiammatorie in Messico e Sud America; diminuisce i mediatori dell'infiammazione, la perdita capillare alveolare e il danno ossidativo in modo dipendente da Nrf2 per alleviare il danno polmonare indotto dalla ventilazione e ripristinare la normale morfologia polmonare [131]. Altri composti vegetali, come l'epigallocatechina gallato, il sulforafano, il resveratrolo, il licopene e l'estratto di tè verde hanno effetti terapeutici sulle malattie infiammatorie attraverso la via di segnalazione Nrf2 [132], [133], [134]. Recentemente un altro fitochimico, eriodictyol, che è presente negli agrumi, è stato segnalato per avere effetti antinfiammatori e antiossidanti sul danno renale indotto da cisplatino e sul danno polmonare acuto indotto da sepsi regolando Nrf2, inibendo NF-? B e inibendo il espressione di citochine nei macrofagi [135], [136]. Tuttavia, numerosi fitochimici mostrano grandi promesse per la prevenzione e il trattamento di varie malattie umane e alcuni sono già entrati nella fase di sperimentazione clinica (Tabella 2).
Questi composti vegetali attivano la via di segnalazione Nrf2 principalmente sotto forma di materiali elettrofili che modificano i residui di cisteina di Keap1, portando al legame nucleare libero Nrf2 con l'ARE, con conseguente attivazione della trascrizione del gene corrispondente.
Sulforafano e suoi effetti su cancro, mortalità, invecchiamento, cervello e comportamento, malattie cardiache e altro
Gli isotiocianati sono alcuni dei composti vegetali più importanti che si possono ottenere nella dieta. In questo video faccio il caso più completo per loro che sia mai stato fatto. Soglia di attenzione breve? Passa al tuo argomento preferito facendo clic su uno dei seguenti punti temporali. Timeline completa di seguito.
Sezioni chiave:
00: 01: 14 - Cancro e mortalità
00: 19: 04 - Invecchiamento
00: 26: 30 - Cervello e comportamento
00: 38: 06 - Riassunto finale
00: 40: 27 - Dose
Timeline completa:
00: 00: 34 - Introduzione di sulforaphane, uno degli obiettivi principali del video.
00: 01: 14 - Consumo di verdure crocifere e riduzione della mortalità per tutte le cause.
00: 02: 12 - Rischio di cancro alla prostata.
00: 02: 23 - Rischio di cancro alla vescica.
00: 02: 34 - Carcinoma polmonare nei fumatori.
00: 02: 48 - Rischio di cancro al seno.
00: 03: 13 - Ipotetico: cosa succede se hai già un cancro? (Interventistica)
00: 03: 35 - Meccanismo plausibile che guida i dati associativi sul cancro e sulla mortalità.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Prova animale che mostra un forte effetto dell'estratto di germogli di broccolo sullo sviluppo del tumore della vescica nei ratti.
00: 06: 06 - Effetto dell'integrazione diretta di sulforafano nei pazienti affetti da cancro alla prostata.
00: 07: 09 - Bioaccumulo di metaboliti di isotiocianato nel tessuto mammario effettivo.
00: 08: 32 - Inibizione delle cellule staminali del carcinoma mammario.
00: 08: 53 - Lezione di storia: le brassiche sono state istituite come aventi proprietà sanitarie anche nell'antica Roma.
00: 09: 16 - La capacità del Sulforaphane di potenziare l'escrezione di cancerogeno (benzene, acroleina).
00: 09: 51 - NRF2 come interruttore genetico tramite elementi di risposta antiossidante.
00: 10: 10 - Come l'attivazione di NRF2 migliora l'escrezione di cancerogeno tramite glutatione-S-coniugati.
00: 10: 34 - I cavoletti di Bruxelles aumentano la glutatione-S-transferasi e riducono il danno al DNA.
00: 11: 20 - La bevanda di germogli di broccoli aumenta l'escrezione di benzene di 61%.
00: 13: 31 - L'omogenato di germogli di broccoli aumenta gli enzimi antiossidanti nelle vie aeree superiori.
00: 15: 45 - Consumo di verdure crocifere e mortalità per malattie cardiache.
00: 16: 55 - La polvere di germogli di broccoli migliora i lipidi nel sangue e il rischio complessivo di malattie cardiache nei diabetici di tipo 2.
00: 19: 04 - Inizio della sezione di invecchiamento.
00: 19: 21 - La dieta arricchita con sulforafano migliora la durata della vita dei coleotteri da 15 a 30% (in determinate condizioni).
00: 20: 34 - L'importanza della bassa infiammazione per la longevità.
00: 22: 05 - Le verdure crocifere e la polvere di germogli di broccoli sembrano ridurre un'ampia varietà di marcatori infiammatori negli esseri umani.
00: 23: 40 - Ricapitolazione di metà video: cancro, sezioni di invecchiamento
00: 24: 14 - Gli studi sui topi suggeriscono che il sulforafano potrebbe migliorare la funzione immunitaria adattativa in età avanzata.
00: 25: 18 - Sulforaphane ha migliorato la crescita dei peli in un modello murino di calvizie. Immagine su 00: 26: 10.
00: 26: 30 - Inizio della sezione cervello e comportamento.
00: 27: 18 - Effetto dell'estratto di germogli di broccoli sull'autismo.
00: 27: 48 - Effetto del glucorapanin sulla schizofrenia.
00: 28: 17 - Inizio della discussione sulla depressione (meccanismo e studi plausibili).
00: 31: 21 - Lo studio del mouse utilizzando 10 diversi modelli di depressione indotta da stress mostra sulforapano in modo simile efficace come fluoxetina (prozac).
00: 32: 00 - Lo studio mostra che l'ingestione diretta di glucorafanina nei topi è altrettanto efficace nel prevenire la depressione dal modello di stress sociale di sconfitta.
00: 33: 01 - Inizio della sezione di neurodegenerazione.
00: 33: 30 - Sulforaphane e malattia di Alzheimer.
00: 33: 44 - Sulforaphane e morbo di Parkinson.
00: 33: 51 - Sulforaphane e la malattia di Hungtington.
00: 34: 43 - Inizio della sezione traumatica di lesioni cerebrali.
00: 35: 01 - Sulforaphane iniettato immediatamente dopo TBI migliora la memoria (studio del mouse).
00: 35: 55 - Sulforaphane e plasticità neuronale.
00: 36: 32 - Sulforaphane migliora l'apprendimento nel modello di diabete di tipo II nei topi.
00: 37: 19 - Distrofia muscolare sulforapano e duchenne.
00: 37: 44 - Inibizione della miostatina nelle cellule muscolari satelliti (in vitro).
00: 38: 06 - Ricapitolazione tardiva: mortalità e cancro, danno al DNA, stress ossidativo e infiammazione, escrezione di benzene, malattie cardiovascolari, diabete di tipo II, effetti sul cervello (depressione, autismo, schizofrenia, neurodegenerazione), via NRF2.
00: 40: 27 - Pensieri sulla determinazione di una dose di germogli di broccoli o sulforafano.
00: 41: 01 - Aneddoti su germinazione a casa.
00: 43: 14 - Sulle temperature di cottura e sull'attività del sulforafano.
00: 43: 45 - Conversione batterica intestinale del sulforafano da glucorafanina.
00: 44: 24 - Gli integratori funzionano meglio se combinati con la mirosinasi attiva delle verdure.
00: 44: 56 - Tecniche di cottura e verdure crucifere.
00: 46: 06 - Isotiocianati come goitrogens.
Conclusioni
Attualmente, molte ricerche si sono concentrate sul ruolo della via di segnalazione Nrf2 / Keap1 / ARE nell'infiammazione. Tra gli enzimi sovraregolati da Nrf2, HO-1 è uno degli enzimi rappresentativi della risposta allo stress. HO-1 ha proprietà antinfiammatorie e antiossidanti prominenti. In generale, la via di segnalazione Nrf2 regola anche negativamente le citochine, i fattori di rilascio delle chemochine, le MMP e altri mediatori infiammatori della produzione di COX-2 e iNOS, che influenzano direttamente o indirettamente le relative vie NF-kB e MAPK e altre reti che controllano l'infiammazione. Si suggerisce che le vie di segnalazione Nrf2 e NF-? B interagiscano per regolare la trascrizione o la funzione delle proteine bersaglio a valle. La soppressione o l'inattivazione dell'attività trascrizionale mediata da NF-? B attraverso Nrf2 si verifica molto probabilmente nella fase iniziale dell'infiammazione, poiché NF-? B regola la sintesi de novo di una serie di mediatori pro-infiammatori. Tuttavia, ci sono ancora alcune limitazioni nella ricerca come se ci siano connessioni tra Nrf2 e altre vie di segnalazione come JAK / STAT, il significato degli attuali attivatori Nrf2 derivati da fonti vegetali naturali nell'infiammazione e come migliorare l'attività biologica e migliorare il targeting di questi composti. Questi richiedono un'ulteriore convalida sperimentale.
Inoltre, la via di segnalazione Nrf2 può regolare> 600 geni [163], di cui> 200 codificano proteine citoprotettive [164] che sono anche associate a infiammazione, cancro, malattie neurodegenerative e altre malattie importanti [165]. Evidenze crescenti suggeriscono che la via di segnalazione Nrf2 è deregolamentata in molti tumori, con conseguente espressione aberrante della batteria genica dipendente da Nrf2. Inoltre, l'infiammazione gioca un ruolo importante nelle malattie legate allo stress ossidativo, specialmente nel cancro. L'applicazione di diversi attivatori Nrf2 per contrastare l'infiammazione può provocare un'espressione aberrante dei geni a valle Nrf2 che induce oncogenesi e resistenza alla chemio e / o radioterapia. Pertanto, attivatori altamente specifici di Nrf2 possono essere sviluppati per minimizzare i suoi effetti pleiotropici. Diversi attivatori di Nrf2 hanno mostrato un significativo miglioramento delle funzioni antinfiammatorie nelle malattie legate allo stress ossidativo. Il miglior esempio di attivatore Nrf2 approvato dalla FDA e ampiamente utilizzato per il trattamento di malattie infiammatorie come la sclerosi multipla (SM) è il dimetilfumarato. Tecfidera (nome registrato di dimetilfumarato da Biogen) utilizzato efficacemente per trattare forme recidivanti di sclerosi multipla in un gran numero di pazienti [152]. Tuttavia, l'efficacia dell'utilizzo degli attivatori Nrf2 per il trattamento delle malattie infiammatorie richiede un'ulteriore convalida per evitare gli effetti deleteri di Nrf2. Pertanto, lo sviluppo di terapie per l'attività antinfiammatoria mediata da Nrf2 potrebbe avere un impatto clinico significativo. Gli studi in corso sul percorso di segnalazione Nrf2 in tutto il mondo sono dedicati allo sviluppo di agenti terapeutici altamente mirati per controllare i sintomi dell'infiammazione e per prevenire e curare il cancro, nonché le malattie neurodegenerative e altre importanti malattie.
In conclusione, Nrf2 rileva i livelli di stress ossidativo nel corpo umano e in definitiva aiuta a promuovere la regolazione di enzimi e geni antiossidanti e disintossicanti. Poiché l'infiammazione cronica causata da un aumento dei livelli di stress ossidativo è stata associata a malattie neurodegenerative, Nrf2 può svolgere un ruolo essenziale nel trattamento di problemi di salute come il morbo di Alzheimer, tra gli altri. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
Discussione aggiuntiva sull'argomento: alleviare il dolore al ginocchio senza chirurgia
Il dolore al ginocchio è un sintomo ben noto che può verificarsi a causa di una varietà di lesioni al ginocchio e / o condizioni, tra cui lesioni sportive. Il ginocchio è una delle articolazioni più complesse del corpo umano in quanto è costituito dall'intersezione di quattro ossa, quattro legamenti, vari tendini, due menischi e cartilagine. Secondo l'American Academy of Family Physicians, le cause più comuni di dolore al ginocchio includono sublussazione rotulea, tendinite rotulea o ginocchio del saltatore e malattia di Osgood-Schlatter. Sebbene il dolore al ginocchio sia più probabile che si verifichi nelle persone di età superiore ai 60 anni, il dolore al ginocchio può verificarsi anche nei bambini e negli adolescenti. Il dolore al ginocchio può essere trattato a casa seguendo i metodi RICE, tuttavia, gravi lesioni al ginocchio possono richiedere cure mediche immediate, compresa la cura chiropratica.
Le malattie neurodegenerative, come il morbo di Alzheimer e il morbo di Parkinson, colpiscono milioni di persone in tutto il mondo. Sono disponibili varie opzioni di trattamento per trattare i sintomi di diverse malattie neurodegenerative, sebbene i risultati siano spesso limitati. Studi di ricerca hanno trovato che lo stress ossidativo causato da fattori sia interni che esterni può essere una causa per lo sviluppo di malattie neurodegenerative. Il fattore di trascrizione, Nrf2, è stato determinato a funzionare come un importante meccanismo di difesa contro lo stress ossidativo. Lo scopo dell'articolo qui sotto è quello di mostrare gli effetti di Nrf2 su malattie neurodegenerative.
Modulazione di Proteostasi mediante Fattore di trascrizione NRF2
Le malattie neurodegenerative sono legate all'accumulo di specifici aggregati proteici, suggerendo una connessione intima tra il cervello ferito e la perdita di proteostasi. La proteostasi si riferisce a tutti i processi attraverso i quali le cellule controllano l'abbondanza e il ripiegamento del proteoma grazie ad un'ampia rete che integra la regolazione delle vie di segnalazione, dell'espressione genica e dei sistemi di degradazione delle proteine. Questa recensione tenta di riassumere i risultati più rilevanti sulla modulazione trascrizionale della proteostasi esercitata dal fattore di trascrizione NRF2 (fattore 2 simile a un fattore nucleare (2 derivato da eritroidi). NRF2 è stato classicamente considerato il principale regolatore della risposta delle cellule antiossidanti, sebbene attualmente stia emergendo come componente chiave dei macchinari di trasduzione per mantenere la proteostasi. Come discuterà, NRF2 potrebbe essere immaginato come un hub che compila segnali di emergenza derivati dall'accumulazione di proteine misfolded al fine di costruire una risposta trascrizionale coordinata e perdurabile. Ciò è ottenuto dalle funzioni di NRF2 relative al controllo dei geni coinvolti nel mantenimento della fisiologia del reticolo endoplasmatico, del proteasoma e dell'autofagia.
parole chiave:Malattie neurodegenerative, risposta proteica non spiegata, proteasoma, ubiquitina, autofagia, stress ossidativo
Fattore Nucleare (2 derivato da eritroide) 2 (NRF2) è una proteina base-leucina-cerniera considerata al giorno d'oggi come un maestro regolatore dell'omeostasi cellulare. Controlla l'espressione basale e stress-inducibile di oltre i geni 250 che condividono in comune un potenziatore cis-acting denominato elemento di risposta antiossidante (ARE) [1], [2], [3], [4], [5]. Questi geni partecipano alle reazioni di disintossicazione di fase I, II e III, al glutatione e al metabolismo di peroxiredoxin / thioredoxin, alla produzione di NADPH attraverso la via del pentoso fosfato e l'enzima malico, l'ossidazione degli acidi grassi, il metabolismo del ferro e la proteostasi [6]. Date queste ampie funzioni citoprotettive, è possibile che un singolo colpo farmacologico in NRF2 possa mitigare l'effetto dei principali colpevoli di malattie croniche, tra cui lo stress ossidativo, infiammatorio e proteotossico. Il ruolo di NRF2 nella modulazione della difesa antiossidante e la risoluzione dell'infiammazione sono stati affrontati in numerosi studi (recensione in [7]). Qui, ci concentreremo sul suo ruolo nella proteostasi, cioè il controllo omeostatico della sintesi proteica, del ripiegamento, del traffico e della degradazione. Verranno forniti esempi nel contesto delle malattie neurodegenerative.
La perdita di proteostasi influenza l'attività di NRF2 nelle malattie neurodegenerative
Un segno distintivo generale delle malattie neurodegenerative è il verificarsi di aggregazioni aberranti di alcune proteine. Pertanto, aggregati proteici mal ripiegati di? -Sinucleina (? -SYN) si trovano nelle placche del morbo di Parkinson (PD),? -Amiloide (A?) E grovigli neurofibrillari TAU iperfosforilati nella malattia di Alzheimer (AD), huntingtina (Htt) in Malattia di Huntington (HD), superossido dismutasi 1 (SOD1) e proteina legante il DNA di TAR 43 (TDP-43) nella sclerosi laterale amiotrofica (SLA), proteina prionica (PrP) nelle encefalopatie spongiformi, ecc. Gli aggregati proteici possono avere un impatto su diversi percorsi cellulari, che a loro volta possono influenzare i livelli e l'attività di NRF2.
Diversi livelli di regolazione controllano strettamente l'attività di NRF2
In condizioni fisiologiche, le cellule mostrano bassi livelli di proteina NRF2 a causa del suo rapido turnover. In risposta a diversi stimoli, la proteina NRF2 viene accumulata, entra nel nucleo e aumenta la trascrizione dei geni contenenti ARE. Pertanto, la gestione dei livelli di proteina NRF2 è un punto chiave che dovrebbe integrare segnali di input positivi e negativi. Come discuterà ulteriormente, NRF2 è attivato da diversi meccanismi sovrapposti per orchestrare una risposta rapida ed efficiente ma d'altra parte NRF2 potrebbe essere inibito, probabilmente in una seconda fase, al fine di disattivare la sua risposta.
Dal punto di vista classico, l'attivazione di NRF2 è stata considerata come una conseguenza della risposta cellulare a composti ossidanti o elettrofili. A questo proposito, l'adattatore della ligasi E3 dell'ubiquitina, la proteina 1 associata a Kelch-like ECH (KEAP1) gioca un ruolo cruciale. I dettagli molecolari saranno ulteriormente trattati nella Sezione 4.1. In breve, KEAP1 agisce come un sensore redox a causa dei residui critici di cisteina che portano all'ubiquitinazione di NRF2 e alla degradazione proteasomica. Oltre a questa classica modulazione, NRF2 è profondamente regolato dalla segnalazione di eventi. In effetti, è stato dimostrato che diverse chinasi fosforilano e regolano NRF2. Ad esempio, NRF2 può essere fosforilato dalle protein chinasi attivate dal mitogeno (MAPK), sebbene il suo contributo all'attività di NRF2 rimanga poco chiaro [8], [9], [10], [11]. Chinasi PKA così come alcuni isoenzimi PKC [12], CK2 [13] o Fyn [14] fosforilano NRF2 modificandone la stabilità. Un precedente lavoro del nostro gruppo ha riportato che glicogeno sintasi kinse-3? (GSK-3?) Inibisce NRF2 per esclusione nucleare e degradazione proteasomiale [15], [25], [26], [27], [28], [29], [30]. I dettagli molecolari saranno discussi nella Sezione 4.1. Inoltre, NRF2 è sottoposto ad altri tipi di regolamentazione. Ad esempio, l'acetilazione di NRF2 da parte di CBP / p300 aumenta la sua attività [17], mentre è inibita da miR153, miR27a, miR142-5p e miR144 [16] o dalla metilazione delle isole citosina-guanina (CG) all'interno del promotore NRF2 [18].
Impatto degli aggregati proteici sui meccanismi regolatori di NRF2
In questa sezione ci concentreremo su come l'accumulo di proteine misfolded potrebbe avere un impatto sull'attività di NRF2 fornendo alcuni dei percorsi sopra menzionati come esempi illustrativi. In primo luogo, dobbiamo considerare che l'accumulo di proteine è strettamente legato al danno ossidativo. Infatti, l'accumulo e l'aggregazione di proteine mal ripiegate inducono una produzione anormale di specie reattive dell'ossigeno (ROS) dai mitocondri e da altre fonti [19]. Come accennato in precedenza, ROS modificherà le cisteine sensibili al redox di KEAP1 portando al rilascio, stabilizzazione e localizzazione nucleare di NRF2.
Per quanto riguarda le proteinopatie, un esempio di eventi di segnalazione disregolati che possono influenzare NRF2 è fornito dall'iperattivazione di GSK-3? in AD. GSK-3?, Noto anche come chinasi TAU, partecipa alla fosforilazione di questa proteina associata ai microtubuli, con conseguente aggregazione, formazione di grovigli neurofibrillari e interruzione del trasporto assonale (rivisto in [20]). D'altra parte, GSK-3? riduce drasticamente i livelli e l'attività di NRF2 come menzionato sopra. Sebbene non ampiamente accettata, la cascata dell'amiloide propone che tossico A? gli oligomeri aumentano GSK-3? attività insieme all'iperfosforilazione della TAU e alla morte dei neuroni [21], [22]. Esistono diversi modelli per spiegare come A? favorisce GSK3-? attività. Ad esempio, A? si lega al recettore dell'insulina e inibisce le vie di segnalazione PI3K e AKT, che sono cruciali per mantenere GSK-3? inattivato dalla fosforilazione nel suo residuo Ser9 N-terminale [23]. D'altra parte, extracellulare A? interagisce con i recettori Frizzled, bloccando la segnalazione WNT [24] e di nuovo determinando il rilascio di GSK-3 attivo ?. In sintesi, A? l'accumulo porta ad un'iperattivazione anormale di GSK-3?, compromettendo così un'appropriata risposta NRF2.
Come discusso nella sezione seguente, le proteine mal ripiegate portano all'attivazione di PERK e MAPK, che a loro volta regolano su NRF2 [31], [8], [9], [10], [11]. Inoltre, l'attività CBP / p300 disregolata è stata riportata in diverse proteinopatie [32] e una diminuzione globale della metilazione del DNA nei cervelli AD è stata anche dimostrata [33], fornendo quindi motivi per esplorare la rilevanza di questi risultati nel regolamento NRF2.
Noi e altri abbiamo osservato in necroscopie di pazienti con PD e AD un aumento dei livelli di proteina NRF2 e alcuni dei suoi bersagli, come l'ossigenoterasi 1 (HMOX1), il NADPH chinone ossidasi 1 (NQO1), p62, ecc., Sia da immunoblot che per immunoistochimica [34], [35], [36], [37], [38], [39]. L'up-regulation di NRF2 in queste malattie è interpretato come un tentativo infruttuoso del cervello malato di recuperare i valori omeostatici. Tuttavia, un altro studio ha indicato che NRF2 è prevalentemente localizzato nel citoplasma dei neuroni dell'ippocampo AD, suggerendo una ridotta attività trascrizionale di NRF2 nel cervello [40]. È concepibile che la disparità di queste osservazioni sia correlata ai cambiamenti nei fattori che controllano NRF2 lungo le fasi progressive della neurodegenerazione.
Tre sistemi principali contribuiscono alla proteostasi, vale a dire la risposta proteica dispiegata (UPR), il sistema di proteasoma ubiquitina (UPS) e l'autofagia. Successivamente, presentiamo prove per immaginare NRF2 come un hub che collega segnali di emergenza attivati da aggregati proteici con il meccanismo derivato dalle proteine.
NRF2 partecipa alla risposta proteica non spiegata (UPR)
Attivazione NRF2 in risposta a UPR
Il ripiegamento delle proteine ossidative nel pronto soccorso è guidato da una serie di percorsi distinti, il più conservato dei quali coinvolge la proteina disolfuro-isomerasi (PDI) e la sulfidril ossidasi ossidoreduttina endoplasmatica 1 (ERO1? E ERO1? Nei mammiferi) come donatore di disolfuro. In breve, la PDI catalizza la formazione e la rottura dei legami disolfuro tra i residui di cisteina all'interno delle proteine, man mano che si ripiegano, a causa della riduzione e dell'ossidazione dei propri amminoacidi cisteina. La PDI viene riciclata dall'azione dell'enzima di mantenimento della casa ERO1, che reintroduce i legami disolfuro nella PDI [41]. L'ossigeno molecolare è l'accettore di elettroni terminale di ERO1, che genera quantità stechiometriche di perossido di idrogeno per ogni legame disolfuro prodotto [42]. Le perossidasi (PRX4) e le perossidasi di glutatione (GPX7 e GPX8) sono enzimi chiave per ridurre il perossido di idrogeno nel pronto soccorso. Quando questo sistema ossidoriduttivo non funziona correttamente, si verifica un accumulo anormale di proteine mal ripiegate nel pronto soccorso e una serie di segnali denominati risposta proteica spiegata (UPR) viene trasmessa al citoplasma e al nucleo per ristabilire l'omeostasi del pronto soccorso [43]. Sono state identificate tre proteine associate alla membrana per rilevare lo stress ER negli eucarioti: fattore di trascrizione attivante 6 (ATF6), ER pancreatico eIF2? chinasi (PERK, anche chinasi ER simile alla proteina chinasi attivata da RNA a doppio filamento) e chinasi1 che richiede inositolo (IRE1). Il dominio luminale di ciascun sensore è legato a una chaperone da 78 kDa chiamata proteina regolata dal glucosio (GRP78 / BIP). Il BIP si dissocia in seguito allo stress ER per legare le proteine dispiegate, portando all'attivazione dei tre sensori [44].
NRF2 e il suo omologo NRF1, anche in relazione alla risposta antiossidante, partecipano alla trasduzione dell'UPR nel nucleo. Nel caso di NRF1, questa proteina si trova nella membrana ER e subisce traslocazione nucleare dopo deglicosilazione o scissione. Quindi, l'attivazione di UPR porta all'elaborazione di NRF1 e all'accumulo nucleare del frammento risultante nel compartimento nucleare. Tuttavia, la capacità di transattivare i geni contenenti ARE di questo frammento NRF1 è ancora in discussione [45].
Glover-Cutter e collaboratori hanno mostrato l'attivazione dell'ortologo NRF2 di C. elegans, SKN-1, con diversi fattori di stress ER. L'aumento dell'espressione di SKN-1 dipendeva da diversi mediatori UPR, inclusi i ortologhi worm IRE1 o PERK [46]. Nelle cellule con carenza di PERK, la sintesi proteica danneggiata porta all'accumulo di perossidi endogeni e conseguente apoptosi [47]. L'effettore utilizzato da PERK per proteggere l'ER da questi perossidi potrebbe essere NRF2, poiché è stato riportato che PERK fosforila NRF2 a Ser40, impedendone così la degradazione da parte di KEAP1 [31]. L'induzione di ASK1 è anche probabile che giochi un ruolo in questa via attraverso l'azione della chinasi mediata da TRAF2 di IRE1 [48]. Sebbene il ruolo dei MAPK nella regolazione di NRF2 sia ancora controverso, è stato recentemente suggerito che il percorso IRE1-TRAF2-ASK1-JNK possa attivare NRF2 [49] (Fig. 1). È interessante notare che in C. elegans e cellule umane, nuove evidenze suggeriscono che la cisteina di solfenilazione della chinasi di IRE1 nel suo ciclo di attivazione inibisce l'UPR mediato da IRE1 e avvia una risposta antiossidante p38 guidata da NRF2. I dati suggeriscono che IRE1 ha un'antica funzione di sentinella citoplasmatica che attiva p38 e NRF2 [50].
Figura 1 Regolamento di NRF2 da parte dell'UPR. L'accumulo di proteine dispiegate o misfoldate all'interno del reticolo endoplasmatico può dare inizio alla risposta proteica dispiegata (UPR). In primo luogo, il chaperone BIP viene rilasciato dal dominio intraluminale dei sensori ER IRE1 e PERK per legare le proteine dispiegate / misfoldate. Ciò consente la dimerizzazione e la trans-auto-fosforilazione dei loro domini citosolici. L'attivazione di PERK provoca la fosforilazione diretta di NRF2 a Ser40, portando alla traslocazione di NRF2 nel nucleo e all'attivazione di geni target. L'attivazione di IRE1 induce il reclutamento di TRAF2 seguito da fosforilazione di ASK1 e JNK e attivazione. Poiché JNK è stato segnalato per fosforilare e attivare NRF2, è ragionevole pensare che l'attivazione di IRE1 porti ad un aumento dell'attività di NRF2.
Molti studi sull'induzione dell'UPR sono stati condotti con l'inibitore della tunicamicina della glicosilazione proteica. NRF2 sembra essere essenziale per la prevenzione della morte cellulare per apoptosi indotta dalla tunicamicina [31] e la sua attivazione in queste condizioni è guidata dalla degradazione autofagica di KEAP1 [51]. Di conseguenza, il silenziamento mediato da shRNA dell'espressione di NRF2 nelle cellule? TC-6, una linea cellulare insulinoma? Murina, ha aumentato significativamente la citotossicità indotta dalla tunicamicina e ha portato ad un aumento dell'espressione del marker di stress ER pro-apoptotico CHOP10. D'altra parte, l'attivazione di NRF2 da parte dell'1,2-ditiolo-3-tione (D3T) ha ridotto la citotossicità della tunicamicina e attenuato l'espressione di CHOP10 e PERK [52]. È interessante notare che i neuroni olfattivi sottoposti ad applicazione sistemica di tunicamicina hanno aumentato NRF2 in parallelo con altri membri UPR come CHOP, BIP, XBP1 [53]. Questi risultati sono stati estesi a studi in vivo, poiché l'infusione ventricolare laterale di tunicamicina nei ratti ha indotto l'espressione di PERK e NRF2 nell'ippocampo accompagnata da deficit cognitivi significativi, aumento della fosforilazione di TAU e depositi di A-42 [54].
NRF2 Up-Regola i geni chiave per il mantenimento della Fisiologia dell'ER
Il lume ER necessita di un'abbondante fornitura di GSH dal citosol per mantenere la chimica del disolfuro. NRF2 modula enzimi cruciali del metabolismo GSH nel cervello, come il trasporto di cistina / glutammato,? -Glutammato cisteina sintetasi (? -GS), glutammato-cisteina ligasi catalitica e subunità modulatrici (GCLC e GCLM), glutatione reduttasi (GR) e glutatione perossidasi (GPX) (rivisto in [55]). La rilevanza di NRF2 nel mantenimento di GSH in ER è supportata dalla scoperta che l'attivazione farmacologica o genetica di NRF2 si traduce in una maggiore sintesi di GSH tramite GCLC / GCLM, mentre l'inibizione dell'espressione di questi enzimi da NRF2-knockdown ha causato un accumulo di danni proteine all'interno del pronto soccorso che portano all'attivazione di UPR [56].
In C. elegans diversi componenti dei geni bersaglio UPR regolati da SKN-1, inclusi Ire1, Xbp1 e Atf6. Sebbene NRF2 regoli l'espressione di diversi geni della perossidasi (PRX) e della glutatione perossidasi (GPX) nei mammiferi (rivisto in [57]), solo GPX8 è un enzima ER localizzato in buona fede, che ospita il segnale di recupero KDEL [58]. La perdita di GPX8 causa l'attivazione di UPR, la fuoriuscita di perossido di idrogeno derivato da ERO1? Nel citosol e la morte cellulare. Perossido di idrogeno derivato da ERO1? l'attività non può diffondersi dal pronto soccorso al citosol a causa dell'azione concertata di GPX8 e PRX4 [59]. A questo proposito, un'analisi della matrice di espressione genica del percorso di difesa antiossidante utilizzando RNA da tessuto di topo wild type e NRF2-null, ha rivelato che l'espressione di GPX8 era down-regolata in assenza di NRF2 [60]. In linea con ciò, l'analisi del trascrittoma da campioni di pazienti affetti da neoplasie mieloproliferative, policitemia o mielofibrosi, malattie associate anche a stress ossidativo e infiammazione cronica di basso grado, mostrano livelli di espressione più bassi sia di NRF2 che di GPX8 rispetto ai soggetti di controllo [61]. Non ci sono ancora studi che coinvolgono specificamente la GPX8 nella protezione del cervello umano, ma un'analisi del trascrittoma nei topi indica un aumento compensatorio della GPX8 in risposta alla tossina parkinsoniana MPTP [62].
Impatto di NRF2 sulla disregolazione UPR nelle malattie neurodegenerative
Il malfunzionamento degli enzimi PDI e l'attivazione cronica dell'UPR potrebbero successivamente avviare o accelerare la neurodegenerazione. Neuroni colpiti dalla malattia, modelli animali di malattie neurodegenerative e tessuti umani post-mortem hanno evidenziato l'up-regulation di diversi marcatori UPR nella maggior parte di questi disturbi. L'alterazione del pathway PDI / UPR nelle malattie neurodegenerative è stata ben rivista in [63] ma i seguenti punti salienti dei campioni post-mortem del cervello dovrebbero essere considerati. I livelli di PDI sono aumentati nei neuroni portatori di tanghi e nei corpi di Lewy dei pazienti con AD e PD, rispettivamente [64], [65]. PDI ed ERP57 sono regolati in alto nel CSF da pazienti con SLA e nel cervello da soggetti con CJD [66], [67], [68]. BIP, PERK, IRE1 e ATF6 sono elevati nei campioni da pazienti con AD, PD o ALS [69], [70], [71], [67]. BIP, CHOP e XBP1 sono elevati nei campioni cerebrali post-mortem da HD [72], [73]. Inoltre, l'up-regulation di ERP57, GRP94 e BIP è stato trovato nei tessuti della corteccia da pazienti con CJD [74]. Complessivamente, questa evidenza rivela che l'accumulo di proteine mal ripiegate nel parenchima cerebrale porta a un'attivazione deleterio e cronica dell'UPR. È interessante notare che c'è uno studio recente che collega l'attivazione di NRF2 con il PERK all'inizio dell'AD. In questo studio, gli autori hanno analizzato se i cambiamenti mediati dallo stress ossidativo in NRF2 e UPR possono costituire eventi precoci nella patogenesi dell'AD utilizzando le cellule del sangue periferico umano e un modello di topo transgenico AD in diverse fasi della malattia. Aumento dello stress ossidativo e aumento di pSer40-NRF2 sono stati osservati nelle cellule mononucleate del sangue periferico umano isolate da individui con compromissione cognitiva lieve. Inoltre, hanno segnalato una compromissione dell'omeostasi del calcio ER e dei marcatori di stress ER sovrastimolati in queste cellule da individui con compromissione cognitiva lieve e lieve AD [75].
Mutua regolamentazione di NRF2 e Ubiquitin Proteasome System (UPS)
L'UPS modula i livelli di proteine NRF2
L'UPS partecipa alla degradazione delle proteine danneggiate o malriposte e controlla i livelli delle molecole regolatrici chiave nel citosol e nel nucleo. Il nucleo centrale di questo sistema è un grande enzima multisubunit che contiene un complesso proteolitico attivo denominato 20S. Il proteasoma core 20S degrada le proteine dispiegate, ma il legame con i diversi complessi proteici regolatori ne modifica la specificità e l'attività del substrato. Ad esempio, l'aggiunta di una o due subunità regolatorie 19S al core 20S costituisce il proteasoma 26S e cambia la sua specificità rispetto alle proteine native piegate [76], [77]. La degradazione del proteasomal ha bisogno del legame covalente dell'ubiquitina. La coniugazione dell'ubiquitina procede attraverso un meccanismo a cascata in tre fasi. Innanzitutto, l'enzima che attiva l'ubiquitina E1 attiva l'ubiquitina in una reazione che richiede ATP. Quindi, un enzima E2 (ubiquitina-carrier protein o ubiquitin-conjugating enzyme) trasferisce l'ubiquitina attivata da E1 al substrato che è specificamente legato a un membro della famiglia delle ligasi della proteina ubiquitina, chiamata E3. Sebbene il destino esatto della proteina ubiquitinata dipenda dalla natura della catena dell'ubiquitina, questo processo generalmente provoca la degradazione del proteasoma 26S [78].
L'E3-ligasi KEAP1 è il più noto inibitore di NRF2. Il meccanismo del regolamento KEAP1 spiega elegantemente come i livelli di NRF2 si adattano alle fluttuazioni ossidanti. In condizioni basali, NRF2 recentemente sintetizzato viene afferrato dall'omodimero KEAP1, che lega una molecola NRF2 a due sequenze di ammino acidi con affinità bassa (aspartato, leucina, glicina; DLG) e alta (glutammato, treonina, glicina, glutammato, ETGE). L'interazione con KEAP1 aiuta a presentare NRF2 al complesso proteico CULLIN3 / RBX1, determinando la sua ubiquitinazione e la conseguente degradazione del proteasomal. Tuttavia, la modifica redox di KEAP1 impedisce la presentazione di NRF2 all'UPS rappresentato da CULLIN3 / RBX1. Di conseguenza, NRF2 appena sintetizzato sfugge alla degradazione dipendente da KEAP1, si accumula nel nucleo e attiva i geni contenenti ARE [79], [80], [81], [82].
L'adattatore E3-ligasi? -TrCP è anche un omodimero che partecipa agli eventi di segnalazione relativi alla fosforilazione di NRF2 da parte di GSK-3 ?. Questa chinasi fosforila residui di serina specifici di NRF2 (aspartato, serina, glicina, isoleucina serina; DSGIS) per creare un dominio di degradazione che viene quindi riconosciuto da? -TrCP e contrassegnato per la degradazione del proteasoma da un complesso CULLIN1 / RBX1. L'identificazione degli amminoacidi specifici che sono fosforilati da GSK-3? in questo degron è stata condotta una combinazione di mutagenesi sito-diretta del dominio Neh6, elettroforesi su gel 2D [15], [26] e spettroscopia di massa [83]. Di conseguenza, l'inibizione di GSK-3? da farmaci altamente selettivi o siRNA contro le isoforme GSK-3 ha determinato un aumento dei livelli di proteina NRF2. Risultati simili sono stati trovati con siRNA contro le isoforme di? -TrCP 1 e 2. Stabilizzazione di NRF2 dopo GSK-3? l'inibizione si è verificata in fibroblasti embrionali di topo KEAP1-carenti e in un mutante di delezione NRF2 espresso ectopicamente privo dei residui ETGE critici per il legame ad alta affinità con KEAP1, dimostrando ulteriormente una regolazione indipendente da KEAP1.
Nel contesto delle malattie neurodegenerative, possiamo prevedere la modulazione di NRF2 da parte dell'UPS in due modi diversi. Da un lato, il sistema KEAP1 rileverebbe uno squilibrio redox derivato dall'accumulo di proteine mal ripiegate, mentre l'asse GSK-3 /? - TrCP agirebbe come un partecipante attivo nella trasduzione del segnale alterata dalla perdita di proteostasi (Fig.2).
Figura 2 L'UPS controlla strettamente i livelli NRF2. In condizioni omeostatiche, bassi livelli di NRF2 sono mantenuti dall'azione degli adattatori delle ligasi E3 KEAP1 e? -TrCP. A sinistra, NRF2 si lega ai domini Kelch di un omodimero KEAP1 attraverso motivi di affinità bassa (DLG) e alta (ETGE). Attraverso il suo dominio BTB, KEAP1 si lega simultaneamente a un complesso CULLIN3 / RBX1, consentendo l'ubiquitinazione e la degradazione di NRF2 da parte del proteasoma 26 S. Inoltre, GSK-3? fosforila i residui di Ser335 e Ser338 di NRF2 per creare un dominio di degradazione (DpSGIpSL) che viene poi riconosciuto dall'adattatore dell'ubiquitina ligasi? -TrCP e contrassegnato per la degradazione del proteasoma da un complesso CULLIN3 / RBX1. A destra, dopo l'esposizione a specie reattive dell'ossigeno o residui di Cys critici per elettrofili in KEAP1 vengono modificati, rendendo KEAP1 incapace di interagire in modo efficiente con NRF2 o CULLIN3 / RBX1 e quindi questo fattore di trascrizione aumenta la sua emivita e l'attività trascrizionale nei confronti dei geni ARE. Le vie di segnalazione che provocano l'inibizione di GSK-3 ?, tale fosforilazione di AKT a Ser9, provocano una degradazione alterata di NRF2 da parte del proteasoma, accumulo e induzione di geni bersaglio.
NRF2 aumenta l'attività dell'UPS attraverso il controllo trascrizionale delle subunità proteasiche
NRF2 regola l'espressione di diverse subunità del proteosoma, proteggendo così la cellula dall'accumulo di proteine tossiche. Venti geni correlati al proteasoma e all'ubiquitinazione sembrano essere regolati da NRF2, secondo un'ampia analisi di microarray da RNA epatico che è stata impostata con l'induttore NRF2 D3T [84]. In uno studio posteriore, gli stessi autori hanno evidenziato che l'espressione della maggior parte delle subunità del proteasoma 26S era aumentata fino a tre volte nel fegato di topi trattati con D3T. I livelli di proteine della subunità e l'attività del proteasoma sono stati aumentati in modo coordinato. Tuttavia, nessuna induzione è stata osservata nei topi in cui il fattore di trascrizione NRF2 è stato interrotto. L'attività del promotore della subunità proteasoma PSMB5 (20S) è aumentata con la sovraespressione di NRF2 o il trattamento con attivatori nei fibroblasti embrionali di topo e le ARE sono state identificate nel promotore prossimale di PSMB5 [85]. L'attivazione farmacologica di NRF2 ha portato a livelli di espressione elevati di subunità rappresentative del proteasoma (PSMA3, PSMA6, PSMB1 e PSMB5) solo in fibroblasti umani non senescenti contenenti NRF2 funzionale [86]. L'attivazione di NRF2 durante l'adattamento allo stress ossidativo determina un'elevata espressione di PSMB1 (20S) e PA28? subunità (o S11, regolatore del proteasoma) [87]. Inoltre, i risultati delle cellule staminali embrionali umane hanno rivelato che NRF2 controlla l'espressione della proteina di maturazione del proteasoma (POMP), un chaperone del proteasoma, che a sua volta modula la proliferazione di cellule staminali embrionali umane che si auto-rinnovano, la differenziazione dei tre strati germinali e la riprogrammazione cellulare [ 88]. Tutti insieme, questi studi indicano che NRF2 regola l'espressione dei componenti chiave dell'UPS e quindi contribuisce attivamente alla clearance delle proteine che altrimenti sarebbero tossiche.
L'asse NRF2-UPS nelle malattie neurodegenerative
Il ruolo dell'UPS nelle malattie neurodegenerative è un campo di intenso dibattito. Gli studi iniziali hanno riportato una diminuzione dell'attività proteasoma nelle necroscopie umane di pazienti affetti da diverse malattie neurodegenerative. Tuttavia, altri studi che impiegano approcci in vitro e in vivo hanno rilevato un'attività proteasoma invariata o addirittura aumentata (revisionata in [89]). Una possibile spiegazione di questa discrepanza è che i livelli dei componenti dell'UPS potrebbero cambiare durante la progressione della malattia e in diverse regioni del cervello, come è stato suggerito per gli obiettivi NRF2.
Nonostante questa controversia, va notato che l'up-regulation dei geni del proteasoma contenenti ARE rafforzerà l'UPS aumentando la clearance delle proteine tossiche nel cervello. Infatti, l'ablazione di NRF1, anche modulatore della risposta antiossidante, nelle cellule neuronali porta a compromissione dell'attività del proteasoma e della neurodegenerazione. Gli esperimenti di immunoprecipitazione della cromatina e l'analisi trascrizionale hanno dimostrato che PSMB6 è regolato da NRF1. Inoltre, il profilo di espressione genica ha portato all'identificazione di NRF1 come regolatore chiave trascrizionale dei geni del proteasoma nei neuroni, suggerendo che le perturbazioni in NRF1 possono contribuire alla patogenesi delle malattie neurodegenerative [90]. È interessante notare che NRF1 e la sua isoforma lunga denominata TCF11 hanno mostrato di up-regolare i geni del proteasoma contenenti ARE sull'inibizione del proteasoma in un anello di feedback per compensare la ridotta attività proteolitica [91], [92].
Per quanto riguarda NRF2, esiste una correlazione tra la riduzione dei livelli di NRF2, RPT6 (19 S) e PSMB5 (20 S) nel mesencefalo di topi con deficit di DJ-1 trattati con il paraquat della neurotossina [93]. Inoltre, il composto sulforafano (SFN) presente in natura fornisce un'immagine più robusta di NRF2 come un modulatore cruciale dell'UPS. Esperimenti in vitro con neuroblastoma murino Le cellule di Neuro2A hanno evidenziato una maggiore espressione delle subunità catalitiche del proteasoma, nonché le sue attività peptidasi in risposta a SFN. Questo farmaco ha protetto le cellule dalla citotossicità perossido di idrogeno e dall'ossidazione delle proteine in modo dipendente dalla funzione del proteasoma [94]. Inoltre, Liu e collaboratori hanno impiegato un topo reporter per monitorare l'attività dell'UPS in risposta a SFN nel cervello. Questi topi esprimono ubiquitariamente la proteina di fluorescenza verde (GFP) fusa con un segnale di degradazione costitutiva che promuove la sua rapida degradazione da parte dell'UPS (GFPu). Nella corteccia cerebrale, SFN ha ridotto il livello di GFPu con un aumento parallelo delle attività simili a chimotripsina (PSMB5), caspase (PSMB2) e trypsin-like (PSMB1) del proteasoma 20 S. Inoltre, il trattamento delle cellule derivate da Huntington con SFN ha rivelato che l'attivazione di NRF2 ha migliorato la degradazione di mHtt e ridotto la citotossicità di mHtt [95]. Il principale meccanismo dell'azione SFN è attraverso l'induzione di NRF2 [96]. Il contributo specifico di NRF2 deve essere affrontato impiegando sistemi NRF2-null in ulteriori studi.
Connessione funzionale tra NRF2 e Macroautophagy
I livelli delle proteine NRF2 sono modulati dalla proteina dell'adattatore P62
L'autofagia si riferisce alla degradazione dei componenti citosolici all'interno dei lisosomi. Questo processo viene utilizzato per la rimozione di proteine a lunga vita e malriposte e di organelli danneggiati. Un collegamento diretto tra NRF2 e autofagia è stato osservato per la prima volta in relazione alla proteina dell'adattatore p62, anche denominata SQSTM1 [97], [98], [99], [100], [101]. Questa proteina trasferisce le proteine ubiquitinate alle macchine di degradazione proteasomale e lisosomiale e sequestra le proteine danneggiate in aggregati prima della loro degradazione. P62 presenta un dominio UBA (ubiquitin-associated), per il legame alle proteine ubiquitinate e una regione LIR (LC3-interacting) per l'integrazione con la membrana autofagosomica attraverso il recettore dell'autofagia LC3.
Sebbene l'induzione mediata da p62 di NRF2 e dei suoi geni bersaglio sia stata segnalata per la prima volta nel 2007 [102], il meccanismo molecolare non è stato completamente compreso fino alla scoperta della sua interazione con KEAP1 [103], [98], [99], [100 ], [101]. Komatsu e colleghi hanno identificato una regione di interazione KEAP1 (KIR) in p62 che legava KEAP1 nella stessa tasca superficiale di base di NRF2 e con un'affinità di legame simile al motivo ETGE in NRF2, suggerendo una competizione tra p62 e NRF2. La fosforilazione di Ser351 nel motivo KIR in p62 (349-DPSTGE-354) ha dimostrato di aumentare la sua affinità per KEAP1, competendo con il legame NRF2 e consentendo il suo accumulo e l'attivazione trascrizionale dei suoi geni bersaglio [98], [99]. Infatti, la sovraespressione di p62 ha portato alla riduzione dell'ubiquitinazione di NRF2 e alla conseguente stabilizzazione, nonché all'induzione dei suoi geni bersaglio [104]. È stato suggerito che alcune chinasi partecipino alla fosforilazione di p62. Il bersaglio dei mammiferi del complesso rapamicina 1 (mTORC1) può essere implicato, poiché il trattamento con l'inibitore di mTOR rapamicina ha soppresso la fosforilazione di p62 e la sottoregolazione di KEAP1 dopo il trattamento con arsenite. Recentemente, è stato dimostrato che TGF -? - chinasi 1 attivata (TAK1) potrebbe anche fosforilare p62, migliorando la degradazione di KEAP1 e la regolazione di NRF2. Gli autori di questo studio suggeriscono che questo sia un modo per regolare la redoxtasis cellulare in condizioni di stato stazionario, poiché la carenza di TAK1 aumenta la regolazione dei ROS in assenza di qualsiasi ossidante esogeno in diversi tessuti di topo in parallelo con una riduzione dei livelli di proteina NRF2 [105 ].
Un costrutto p62 privo del dominio UBA era ancora in grado di legare KEAP1, implicando che l'interazione non dipendesse da KEAP1 ubiquitinato [101]. Tuttavia, l'omologo di p62 in Drosophila melanogaster, denominato Ref (2), non contiene un motivo KIR e non interagisce direttamente con DmKEAP1, sebbene possa legarsi a DmKEAP1 ubiquitinato attraverso il dominio UBA. Inoltre, DmKEAP1 può interagire direttamente con Atg8 (omologo ai mammiferi LC3). Il deficit di KEAP1 risulta in Atg8 e l'induzione dell'autofagia dipendente dall'ortologo di NRF2 CncC e indipendente da TFEB / MITF [106]. La relazione tra NRF2 e autofagia sembra essere conservata, evidenziando la sua rilevanza funzionale.
L'induzione di NRF2 da parte di p62 è il risultato di entrambe le gare per legare KEAP1 e la degradazione di KEAP1 nel lisosoma. Il silenziamento di p62 con siRNA ha raddoppiato l'emivita di KEAP1 in parallelo con una diminuzione di NRF2 e dei suoi geni bersaglio [101]. In accordo, l'ablazione dell'espressione di p62 ha evidenziato un aumento dei livelli di KEAP1 rispetto ai topi wild type. Molto rilevante, l'incremento dei livelli di KEAP1 non è stato influenzato dagli inibitori del proteasoma, ma è stato ridotto con l'autophagy inducente la fame [107]. Infatti, KEAP1 è presente nelle cellule di mammifero in vescicole autofagiche decorate con p62 e LC3 [99], [100], [103]. Tutti questi dati suggeriscono che KEAP1 è un substrato del macchinario di macroautofagia, ma questo problema dovrebbe essere analizzato con maggiori dettagli a causa dell'esistenza di alcuni risultati controversi. I livelli di proteina KEAP1 sono aumentati nei topi Atg7 null, un effettore chiave della macroautofagia [107], ma l'inibizione farmacologica della macroautofagia con torin1, E64 / pepstatin o bafilomicina non ha accumulato KEAP1 [107], [100]. Nel complesso, questi risultati suggeriscono che l'aumento dei livelli di p62 sequestra KEAP1 in vacuoli autofagici e probabilmente questi risultati nella degradazione autofagica di KEAP1 permettendo l'attivazione di NRF2 (Fig. 3). Due diversi studi hanno riportato che l'acido sulfinico riduttasi SESTRINS svolge un ruolo importante in questo contesto. SESTRIN 2 interagisce con p62, KEAP1 e RBX1 e facilita la degradazione dipendente da p62 dell'attivazione di KEAP1 e NRF2 dei geni target [108]. Un altro studio ha dimostrato che SESTRIN 2 interagiva con ULK1 e p62, promuovendo la fosforilazione di p62 a Ser403 che ha facilitato il degrado delle proteine del carico incluso KEAP1 [109].
Figura I livelli di 3 NRF2 sono regolati dalla proteina dell'adattatore p62. La fosforilazione di Ser 351 nel motivo KIR di p62 (349-DPSTGE-354) da mTORC1, TAK1 o altre chinasi determina una maggiore affinità per il legame con KEAP1 a causa della somiglianza con il motivo ETGE in NRF2. Di conseguenza, p62 fosforilato sposta NRF2 e lega KEAP1. Il motivo LIR in p62 consente l'interazione con LC3 nella membrana autofagosomica, in modo tale che il complesso p62-KEAP1 venga infine degradato nel lisosoma. Di conseguenza NRF2 è in grado di accumularsi, traslocare nel nucleo e aumentare la trascrizione dei geni contenenti ARE, incluso p62. Questo meccanismo di regolazione fornisce una risposta NRF2 duratura, poiché KEAP1 deve essere nuovamente sintetizzato per inibire l'attività di NRF2.
Modulazione dei geni Macroautophagy di NRF2
NRF2 regola l'espressione di geni rilevanti per la macroautofagia, così come per l'UPR e l'UPS. La prima evidenza proviene da studi in cui è stato dimostrato che l'espressione di p62 è indotta dall'esposizione a elettrofili, ROS e ossido nitrico [110], [111], [112]. Il meccanismo di induzione è stato descritto alcuni anni dopo con la scoperta che p62 contiene un ARE funzionale nel suo promotore genico [99]. In un recente studio, sono state trovate diverse altre ARE funzionali e validate in seguito a analisi bioinformatica e analisi ChIP. Inoltre, i fibroblasti embrionali di topo e i neuroni corticali dei topi knockout di Nrf2 presentavano un'espressione ridotta di p62, che poteva essere salvata con un lentivirus che esprimeva NRF2. Allo stesso modo, il deficit di NRF2 ha ridotto i livelli di p62 nei neuroni feriti da topi ippocampo [36]. Pertanto, è stato suggerito che l'attivazione di NRF2 aumenta i livelli di p62, con conseguente degradazione di KEAP1 e favorendo l'ulteriore stabilizzazione di NRF2 in un ciclo di feedback positivo. Questo meccanismo non canonico di induzione NRF2 richiede cambiamenti nell'espressione genica e potrebbe essere una risposta rilevante allo stress cellulare prolungato.
La proteina di riconoscimento del carico NDP52 ha dimostrato di essere regolata per via trascrizionale da NRF2. NDP52 funziona in modo simile a p62, riconoscendo proteine ubiquitinate e interagendo con LC3 attraverso un dominio LIR, in modo che i carichi siano degradati nei lisosomi. Sono stati trovati cinque ipotetici valori nella sequenza del DNA del promotore Ndp52. Tre di essi sono stati identificati con diversi costrutti mutanti e test ChIP come indispensabili per la trascrizione Ndp2 mediata da NRF52 [113]. Di nota, i livelli di mRNA di Ndp52 sono stati ridotti nell'ippocampo di topi knockout Nrf2. Una di queste sequenze è stata anche validata in uno studio indipendente come ARE [2] regolato da NRF36.
Tuttavia, il ruolo di NRF2 nella modulazione dell'autofagia non è limitato all'induzione di queste due proteine di riconoscimento del carico. Al fine di ottenere una visione più approfondita del ruolo di NRF2 nella modulazione di ulteriori geni correlati all'autofagia, il nostro gruppo ha esaminato il database di immunoprecipitazione della cromatina ENCODE per due proteine, MAFK e BACH1, che legano le ARE regolate da NRF2. Usando uno script generato dalla sequenza ARE di consenso di JASPAR, abbiamo identificato diverse POS putative in molti geni dell'autofagia. Dodici di queste sequenze sono state convalidate come ARE regolate da NRF2 in nove geni autofagici, la cui espressione è stata ridotta nei fibroblasti di embrioni di topo di topi knockout Nrf2, ma potrebbe essere ripristinata da un lentivirus che esprime NRF2. Il nostro studio ha dimostrato che NRF2 attiva l'espressione di alcuni geni coinvolti in diverse fasi del processo autofagico, tra cui l'inizio dell'autofagia (ULK1), il riconoscimento del carico (p62 e NDP52), la formazione di autofagosomi (ATG4D, ATG7 e GABARAPL1), l'allungamento (ATG2B e ATG5) ) e clearance del autososoma (ATG4D). Di conseguenza, il flusso di autofagia in risposta al perossido di idrogeno è stato compromesso quando NRF2 era assente [36].
Rilevanza Dell'espressione Genica Dei Macroautofagia Mediata Da NRF2 Nei Disturbi Neurodegenerativi
L'autofagia difettosa ha dimostrato di svolgere un ruolo importante in diverse malattie neurodegenerative [114] e l'ablazione dell'autofagia porta alla neurodegenerazione nei topi [115], [116]. Topi knockout di Atg7 hanno rivelato che il deficit di autofagia si traduce in un accumulo di p62 in corpi di inclusione ubiquitina-positivi. KEAP1 è stato sequestrato in questi corpi di inclusione, portando alla stabilizzazione di NRF2 e all'induzione dei geni bersaglio [103]. È importante notare che l'accumulo eccessivo di p62 insieme a proteine ubiquitinate è stato identificato nelle malattie neurodegenerative, tra cui AD, PD e ALS [117]. Infatti, i neuroni che esprimono alti livelli di APP o TAU di pazienti con AD hanno espresso anche p62 e NRF2 nucleare, suggerendo il loro tentativo di degradare gli aggregati intraneuronali attraverso l'autofagia [36].
La carenza di NRF2 aggrava l'aggregazione proteica nel contesto dell'AD. Infatti, livelli aumentati di TAU fosforilata e insolubile in sarkosil si trovano nei topi knockout per Nrf2, sebbene non sia stata rilevata alcuna differenza nelle attività della chinasi o della fosfatasi confrontandola con lo sfondo wild-type [113]. È importante sottolineare che NDP52 è stato dimostrato di co-localizzare con TAU nei neuroni murini e l'interazione diretta tra fosfo-TAU e NDP52 è stata dimostrata da esperimenti di co-immunoprecipitazione sia nei topi che nei campioni di AD, indicando il suo ruolo nella degradazione della TAU. È interessante notare che il silenziamento di NDP52, p62 o NRF2 nei neuroni ha portato a un aumento della fosfo-TAU [113], [118]. Inoltre, sono stati trovati aggregati APP intraneuronali aumentati nell'ippocampo di topi APP / PS1? E9 quando NRF2 era assente. Ciò era correlato a marcatori di autofagia alterati, tra cui un aumento dei rapporti fosfo-mTOR / mTOR e fosfo-p70S6k / p70S6k (indicativi di inibizione dell'autofagia), livelli aumentati di pre-catepsina D e un numero maggiore di corpi multivesicolari [119]. Nei topi che coesprimono APP umana (V717I) e TAU (P301L), il deficit di NRF2 ha portato ad un aumento dei livelli di TAU totale e fosfo-TAU nella frazione insolubile e ad un aumento degli aggregati di APP intraneuronali, insieme a livelli neuronali ridotti di p62, NDP52, ULK1, ATG5 e GABARAPL1. La co-localizzazione tra la proteina adattatore p62 e APP o TAU è stata ridotta in assenza di NRF2 [36]. Nel complesso, questi risultati evidenziano l'importanza di NRF2 nell'autofagia neuronale.
I diversi fattori di trascrizione agiscono coordinatamente per modulare la proteostasi
In condizioni di stato stazionario, la proteostasi viene controllata tramite interazioni proteina-proteina e modificazioni post-traduzionali ottenendo una risposta rapida. Tuttavia, l'adattamento cellulare richiede la regolazione trascrizionale dei geni UPR, UPS e autofagia. Considerando che le cellule nervose sono continuamente sottoposte a insulti tossici di basso grado, tra cui lo stress ossidativo e proteotossico, un rinforzo della proteostasi indotta dalla modulazione trascrizionale potrebbe aiutare a prevenire la degenerazione del cervello.
Nel caso dell'UPR, l'attivazione di ciascuna delle tre braccia porterà infine all'induzione trascrizionale di alcuni geni (rivisti in [43]). Ad esempio, un frammento derivato da ATF6 (ATF6f) si lega agli elementi di risposta allo stress ER (ERSE) e induce l'espressione di diversi geni, tra cui XBPI, BIP e CHOP. Inoltre, la segnalazione PERK porta all'attivazione del fattore di trascrizione ATF4, che controlla l'espressione di più geni correlati a UPR e alcuni altri tra cui i geni bersaglio NRF2 Hmox1 e p62. Infine, l'attivazione di IRE1 determina la generazione di un fattore di trascrizione attivo XBP1 (XBP1s), che controlla la trascrizione dei geni che codificano le proteine coinvolte nel ripiegamento delle proteine.
D'altra parte, NRF1 si è dimostrato necessario per l'espressione genica del proteasomal nel cervello, in quanto i topi knockout Nrf1 hanno esibito un'espressione ridotta di geni che codificano varie subunità del core 20S, così come il complesso regolatore 19S insieme a funzionalità proteasomica compromessa [90 ]. Sia NRF1 che NRF2 si legano alle sequenze ARE nelle regioni del promotore dei suoi geni bersaglio, il che suggerisce che hanno attività trascrizionali sovrapposte, sebbene differiscano nei loro meccanismi regolatori e nella localizzazione cellulare [120].
I fattori di trascrizione della famiglia Forkhead box O (FOXO) controllano l'espressione di più geni correlati all'autofagia. Simile a ciò che avviene con NRF2, ci sono più livelli di regolazione dell'attività dei membri FOXO, che possono essere indotti su stress nutrizionale o ossidativo [121]. Infine, il fattore di trascrizione TFEB, considerato il principale regolatore della biogenesi lisosomiale, svolge un ruolo cruciale nella regolazione dell'autofagia in condizioni di stress nutrizionale. Pertanto, l'inibizione di mTORC1 porta alla traslocazione nucleare di TFEB e all'induzione dell'espressione di geni autofagici [122].
Nel complesso, l'esistenza di diversi regolatori trascrizionali di questi macchinari suggerisce anche diafonia e meccanismi parzialmente ridondanti che possono assicurare la proteostasi in diverse circostanze. Di conseguenza, NRF2 può avere un ruolo rilevante nei tessuti che supportano alti livelli di stress ossidativo. Ad esempio, NRF2 indotto da stress ossidativo può funzionare in condizioni ricche di sostanze nutritive per autophagy sovraregolare in modo trascrizionale, simile a quanto è stato trovato per TFEB in condizioni di fame. Inoltre, il cervello funziona in gran parte sotto condizioni nutritive, ponendo NRF2 come un meccanismo rilevante per attivare l'autofagia nei neuroni.
Promettente Potenziale Terapeutico per NRF2 nelle Proteinopatie
Negli ultimi anni sono stati compiuti notevoli progressi nella conoscenza dei ruoli normativi dell'UPR, dell'UPS e dell'autofagia sull'attività di NRF2, nonché della trascrizione reciproca mediata da NRF2 di componenti di questi tre sistemi. Pertanto, possono sorgere nuove possibilità terapeutiche basate sullo sfruttamento di NRF2 come regolatore cruciale della clearance delle proteine nelle malattie neurodegenerative.
Tuttavia, una questione chiave che rimane è se sarà utile o deleterio aumentare i livelli di NRF2 nel cervello. L'analisi dei dati epidemiologici può fornire una risposta parziale, in quanto indica che il gene NFE2L2 è altamente polimorfico e alcuni polimorfismi a singolo nucleotide trovati nella sua regione regolatrice del promotore possono fornire una gamma di variabilità `` fisiologica '' nell'espressione genica a livello di popolazione e alcuni aplotipi erano associati a una riduzione del rischio e / o all'insorgenza ritardata di AD, PD o SLA [123]. Inoltre, come discusso da Hayes e colleghi [124], l'effetto NRF2 potrebbe avere una risposta a forma di U, il che significa che livelli troppo bassi di NRF2 possono provocare una perdita di citoprotezione e una maggiore suscettibilità ai fattori di stress, mentre una quantità eccessiva di NRF2 potrebbe disturbare l'equilibrio omeostatico verso uno scenario riduttivo (stress riduttivo), che favorirebbe il misfolding e l'aggregazione delle proteine. Bassi livelli di NRF2 nel cervello supportano l'idea che una leggera sovraregolazione possa essere sufficiente per ottenere un beneficio in condizioni patologiche. Infatti, il ruolo protettivo dell'attivazione farmacologica della clearance della proteina mediata da NRF2 è stato dimostrato in diverse colture cellulari di neurodegenerazione e modelli in vivo.
SFN è un attivatore farmacologico di NRF2 che ha dimostrato di indurre l'espressione genica del proteasoma e dell'autofagia [95], [36]. È interessante notare che Jo e colleghi hanno dimostrato che SFN riduce i livelli di TAU fosforilata e aumenta Beclin-1 e LC3-II, suggerendo che l'attivazione di NRF2 può facilitare la degradazione di questa proteina tossica attraverso l'autofagia [113]. Inoltre, la degradazione di mHtt è stata migliorata con SFN, e questo è stato invertito con l'uso di MG132, indicando la degradazione proteasomica di questa proteina tossica [95]. La degradazione mediata dall'autofagia di TAU fosfo e insolubile è stata segnalata con la fisetina flavonoide organica. Questo composto è stato in grado di indurre l'autofagia promuovendo simultaneamente l'attivazione e la traslocazione nucleare di TFEB e NRF2, insieme ad alcuni dei suoi geni bersaglio. Questa risposta è stata prevenuta dal silenziamento TFEB o NRF2 [125]. Bott e colleghi hanno riportato effetti benefici di un attivatore simultaneo di NRF2, NRF1 e HSF1 sulla tossicità delle proteine nell'atrofia muscolare spinale e bulbare, una malattia neurodegenerativa causata dall'espansione delle ripetizioni CAG codificanti la poliglutamina in cui sono presenti aggregati proteici [126]. Il potenziale di attivazione di NRF2 per il trattamento dei disturbi neurodegenerativi è stato dimostrato con l'approvazione di BG-12, la formulazione orale dell'induttore NRF2 dimetilfumarato (DMF), per il trattamento della sclerosi multipla [127], [128]. Il successo del DMF con malattie autoimmuni con una forte componente infiammatoria suggerisce che le malattie neurodegenerative potrebbero trarre beneficio dal riposizionamento di questo farmaco. In un recente studio preclinico su un modello di? -Sinucleinopatia di PD, la DMF ha dimostrato di essere neuroprotettiva a causa, in parte, della sua induzione di autofagia [129]. Gli studi che riportano gli effetti benefici di NRF2 sulla neurodegenerazione ma non si concentrano sul suo effetto sulla clearance delle proteine sono ancora più abbondanti (per una revisione completa, vedere [7]). Questo è abbastanza rilevante, in quanto evidenzia i molteplici processi dannosi che possono essere simultaneamente presi di mira da un singolo colpo in NRF2, inclusi anche lo stress ossidativo, la neuroinfiammazione o la disfunzione mitocondriale. Tuttavia, sarà necessario un lavoro futuro per determinare definitivamente se l'attivazione farmacologica di NRF2 può essere una strategia valida per facilitare la degradazione delle proteine tossiche nel cervello.
Come spiegato prima, GSK-3 esacerbato? l'attività è stata riportata nelle malattie neurodegenerative ed è stato ipotizzato che la conseguente riduzione di NRF2 possa essere parzialmente responsabile dell'outcome deleterio. In queste condizioni patologiche, gli inibitori GSK-3 potrebbero anche cooperare per aumentare i livelli di NRF2 e la proteostasi. Gli effetti benefici degli inibitori GSK-3 sono stati riportati in diversi modelli di neurodegenerazione e, cosa più interessante, è stato dimostrato che la repressione GSK-3 riduce i livelli di proteine tossiche [130], [131], [132], [133]. Sebbene non siano stati ancora osservati collegamenti diretti tra l'inibizione di GSK-3 e la regolazione trascrizionale di NRF2 dei geni che promuovono la proteostasi, è ragionevole ipotizzare che la sottoregolazione dell'attività GSK-3 comporterebbe un aumento dei livelli di NRF2, che alla fine si tradurrà in un rafforzamento proteostasi.
L'attività trascrizionale di NRF2 e la capacità cellulare di mantenere la proteostasi diminuiscono con l'età, principale fattore di rischio per lo sviluppo di malattie neurodegenerative. È ragionevole pensare che il rafforzamento di NRF2 e, di conseguenza, la proteostasi ritarderebbe almeno l'accumulo di aggregati proteici e la neurodegenerazione. Infatti, il trattamento dei fibroblasti senescenti umani con triterpenoide di acido 18? -Glicirretinico (18? -GA) ha promosso l'attivazione di NRF2, portando all'induzione del proteasoma e a una maggiore durata della vita. Questo studio suggerisce che l'attivazione farmacologica di NRF2 è possibile anche in tarda età [86]. Inoltre, uno studio successivo ha mostrato che questo composto mediava SKN-1 e l'attivazione del proteasoma in C.elegans con effetti benefici sulla progressione dell'AD in modelli di nematodi rilevanti [134].
Tutto sommato, l'induzione mediata da NRF2 di geni correlati alla proteostasi sembra essere benefica in diverse proteinopatie.
Sulforafano e suoi effetti su cancro, mortalità, invecchiamento, cervello e comportamento, malattie cardiache e altro
Gli isotiocianati sono alcuni dei composti vegetali più importanti che si possono ottenere nella dieta. In questo video faccio il caso più completo per loro che sia mai stato fatto. Soglia di attenzione breve? Passa al tuo argomento preferito facendo clic su uno dei seguenti punti temporali. Timeline completa di seguito.
Sezioni chiave:
00: 01: 14 - Cancro e mortalità
00: 19: 04 - Invecchiamento
00: 26: 30 - Cervello e comportamento
00: 38: 06 - Riassunto finale
00: 40: 27 - Dose
Timeline completa:
00: 00: 34 - Introduzione di sulforaphane, uno degli obiettivi principali del video.
00: 01: 14 - Consumo di verdure crocifere e riduzione della mortalità per tutte le cause.
00: 02: 12 - Rischio di cancro alla prostata.
00: 02: 23 - Rischio di cancro alla vescica.
00: 02: 34 - Carcinoma polmonare nei fumatori.
00: 02: 48 - Rischio di cancro al seno.
00: 03: 13 - Ipotetico: cosa succede se hai già un cancro? (Interventistica)
00: 03: 35 - Meccanismo plausibile che guida i dati associativi sul cancro e sulla mortalità.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Prova animale che mostra un forte effetto dell'estratto di germogli di broccolo sullo sviluppo del tumore della vescica nei ratti.
00: 06: 06 - Effetto dell'integrazione diretta di sulforafano nei pazienti affetti da cancro alla prostata.
00: 07: 09 - Bioaccumulo di metaboliti di isotiocianato nel tessuto mammario effettivo.
00: 08: 32 - Inibizione delle cellule staminali del carcinoma mammario.
00: 08: 53 - Lezione di storia: le brassiche sono state istituite come aventi proprietà sanitarie anche nell'antica Roma.
00: 09: 16 - La capacità del Sulforaphane di potenziare l'escrezione di cancerogeno (benzene, acroleina).
00: 09: 51 - NRF2 come interruttore genetico tramite elementi di risposta antiossidante.
00: 10: 10 - Come l'attivazione di NRF2 migliora l'escrezione di cancerogeno tramite glutatione-S-coniugati.
00: 10: 34 - I cavoletti di Bruxelles aumentano la glutatione-S-transferasi e riducono il danno al DNA.
00: 11: 20 - La bevanda di germogli di broccoli aumenta l'escrezione di benzene di 61%.
00: 13: 31 - L'omogenato di germogli di broccoli aumenta gli enzimi antiossidanti nelle vie aeree superiori.
00: 15: 45 - Consumo di verdure crocifere e mortalità per malattie cardiache.
00: 16: 55 - La polvere di germogli di broccoli migliora i lipidi nel sangue e il rischio complessivo di malattie cardiache nei diabetici di tipo 2.
00: 19: 04 - Inizio della sezione di invecchiamento.
00: 19: 21 - La dieta arricchita con sulforafano migliora la durata della vita dei coleotteri da 15 a 30% (in determinate condizioni).
00: 20: 34 - L'importanza della bassa infiammazione per la longevità.
00: 22: 05 - Le verdure crocifere e la polvere di germogli di broccoli sembrano ridurre un'ampia varietà di marcatori infiammatori negli esseri umani.
00: 23: 40 - Ricapitolazione di metà video: cancro, sezioni di invecchiamento
00: 24: 14 - Gli studi sui topi suggeriscono che il sulforafano potrebbe migliorare la funzione immunitaria adattativa in età avanzata.
00: 25: 18 - Sulforaphane ha migliorato la crescita dei peli in un modello murino di calvizie. Immagine su 00: 26: 10.
00: 26: 30 - Inizio della sezione cervello e comportamento.
00: 27: 18 - Effetto dell'estratto di germogli di broccoli sull'autismo.
00: 27: 48 - Effetto del glucorapanin sulla schizofrenia.
00: 28: 17 - Inizio della discussione sulla depressione (meccanismo e studi plausibili).
00: 31: 21 - Lo studio del mouse utilizzando 10 diversi modelli di depressione indotta da stress mostra sulforapano in modo simile efficace come fluoxetina (prozac).
00: 32: 00 - Lo studio mostra che l'ingestione diretta di glucorafanina nei topi è altrettanto efficace nel prevenire la depressione dal modello di stress sociale di sconfitta.
00: 33: 01 - Inizio della sezione di neurodegenerazione.
00: 33: 30 - Sulforaphane e malattia di Alzheimer.
00: 33: 44 - Sulforaphane e morbo di Parkinson.
00: 33: 51 - Sulforaphane e la malattia di Hungtington.
00: 34: 43 - Inizio della sezione traumatica di lesioni cerebrali.
00: 35: 01 - Sulforaphane iniettato immediatamente dopo TBI migliora la memoria (studio del mouse).
00: 35: 55 - Sulforaphane e plasticità neuronale.
00: 36: 32 - Sulforaphane migliora l'apprendimento nel modello di diabete di tipo II nei topi.
00: 37: 19 - Distrofia muscolare sulforapano e duchenne.
00: 37: 44 - Inibizione della miostatina nelle cellule muscolari satelliti (in vitro).
00: 38: 06 - Ricapitolazione tardiva: mortalità e cancro, danno al DNA, stress ossidativo e infiammazione, escrezione di benzene, malattie cardiovascolari, diabete di tipo II, effetti sul cervello (depressione, autismo, schizofrenia, neurodegenerazione), via NRF2.
00: 40: 27 - Pensieri sulla determinazione di una dose di germogli di broccoli o sulforafano.
00: 41: 01 - Aneddoti su germinazione a casa.
00: 43: 14 - Sulle temperature di cottura e sull'attività del sulforafano.
00: 43: 45 - Conversione batterica intestinale del sulforafano da glucorafanina.
00: 44: 24 - Gli integratori funzionano meglio se combinati con la mirosinasi attiva delle verdure.
00: 44: 56 - Tecniche di cottura e verdure crucifere.
00: 46: 06 - Isotiocianati come goitrogens.
Il fattore 2 (NF-E2), derivato da eritroide, derivato da eritroide, è un fattore di trascrizione che regola l'espressione di una varietà di enzimi antiossidanti e disintossicanti. Studi di ricerca hanno anche dimostrato il suo ruolo nel controllo dello stress ossidativo. La maggior parte delle malattie neurodegenerative, come il morbo di Alzheimer e il morbo di Parkinson, sono caratterizzate da stress ossidativo e infiammazione cronica, gli obiettivi comuni di Approcci trattamento Nrf2. Dr. Alex Jimenez DC, CCST Insight
Osservazioni conclusive
Il fattore di trascrizione NRF2 orchestra una risposta proteostatica rilevando e modulando i cambiamenti in UPR, UPS e autofagia (Fig. 4). Di conseguenza, la mancanza di NRF2 ha dimostrato di aggravare la proteinopatia, suggerendo che NRF2 è necessario per una clearance ottimale delle proteine. Tutti insieme, possiamo ipotizzare che NRF2 potrebbe essere un obiettivo terapeutico interessante per le proteinopatie.
Figura 4 NRF2 come hub che collega segnali di emergenza derivati da proteotoxic a una risposta trascrizionale protettiva. L'accumulo di proteine unfolded / misfolded porterà all'attivazione della risposta proteica dispiegata (UPR) nel pronto soccorso. L'attivazione di PERK o MAPK può comportare l'induzione transcriptional di Gpx8 residente con ER e diversi enzimi che regolano i livelli di GSH, fondamentali per assicurare il corretto ripiegamento delle proteine. Gli aggregati proteici inibiscono l'attività del proteasoma (UPS), probabilmente evitando la degradazione di NRF2. NRF2 ha dimostrato di modulare specificamente la trascrizione dei geni Psma3, Psma6, Psmb1, Psmb5 e Pomp. Diverse altre sottounità sono state regolate in modo dipendente da NRF2 in risposta a D3T, probabilmente ingrandendo la lista delle subunità proteasiche regolate da NRF2. L'autofagia è la via principale per la degradazione degli aggregati proteici. L'autofagia regola anche NRF2, collegando questo percorso di degradazione con l'induzione trascrizionale NRF2 di p62, Ndp52, Ulk1, Atg2b, Atg4c, Atg5, Atg7 e Gabarapl1.
Secondo l'articolo precedente, mentre i sintomi delle malattie neurodegenerative possono essere trattati attraverso una varietà di opzioni di trattamento, studi di ricerca hanno dimostrato che l'attivazione di Nrf2 può essere un approccio terapeutico utile. Perché Gli attivatori di Nrf2 prendono di mira ampi meccanismi di malattia, tutte le malattie neurodegenerative possono trarre vantaggio dall'uso del fattore di trascrizione Nrf2. I risultati di Nrf2 hanno rivoluzionato il trattamento delle malattie neurodegenerative. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
Discussione aggiuntiva sull'argomento: alleviare il dolore al ginocchio senza chirurgia
Il dolore al ginocchio è un sintomo ben noto che può verificarsi a causa di una varietà di lesioni al ginocchio e / o condizioni, tra cui lesioni sportive. Il ginocchio è una delle articolazioni più complesse del corpo umano in quanto è costituito dall'intersezione di quattro ossa, quattro legamenti, vari tendini, due menischi e cartilagine. Secondo l'American Academy of Family Physicians, le cause più comuni di dolore al ginocchio includono sublussazione rotulea, tendinite rotulea o ginocchio del saltatore e malattia di Osgood-Schlatter. Sebbene il dolore al ginocchio sia più probabile che si verifichi nelle persone di età superiore ai 60 anni, il dolore al ginocchio può verificarsi anche nei bambini e negli adolescenti. Il dolore al ginocchio può essere trattato a casa seguendo i metodi RICE, tuttavia, gravi lesioni al ginocchio possono richiedere cure mediche immediate, compresa la cura chiropratica.
Lo stress ossidativo è descritto come danno cellulare causato da radicali liberi o molecole instabili, che alla fine possono influire sulla funzione sana. Il corpo umano crea radicali liberi per neutralizzare batteri e virus, tuttavia, fattori esterni come l'ossigeno, l'inquinamento e le radiazioni possono spesso anche produrre radicali liberi. Lo stress ossidativo è stato associato a numerosi problemi di salute.
Lo stress ossidativo e altri fattori di stress attivano meccanismi di protezione interni che possono aiutare a regolare la risposta antiossidante del corpo umano. Nrf2 è una proteina che rileva i livelli di stress ossidativo e consente alle cellule di proteggersi da fattori interni ed esterni. È stato anche dimostrato che Nrf2 aiuta a regolare i geni coinvolti nella produzione di enzimi antiossidanti e geni di risposta allo stress. Lo scopo dell'articolo qui sotto è quello di spiegare il effetti di Nrf2 nel cancro
Astratto
Il pathway Keap1-Nrf2 è il principale regolatore delle risposte citoprotettive allo stress ossidativo ed elettrofilo. Sebbene le vie di segnalazione cellulare attivate dal fattore di trascrizione Nrf2 prevengano l'inizio e la progressione del cancro nei tessuti normali e premaligni, nelle cellule completamente maligne l'attività di Nrf2 fornisce un vantaggio di crescita aumentando la chemioresistenza del cancro e migliorando la crescita delle cellule tumorali. In questa recensione grafica, forniamo una panoramica del percorso di Keap1-Nrf2 e della sua disregolazione nelle cellule tumorali. Riassumiamo anche brevemente le conseguenze dell'attivazione di Nrf2 costitutiva nelle cellule tumorali e come questo possa essere sfruttato nella terapia genica del cancro.
parole chiave:Nrf2, Keap1, Cancro, elemento di risposta antiossidante, terapia genica
Introduzione
La via Keap1-Nrf2 è il principale regolatore delle risposte citoprotettive agli stress endogeni ed esogeni causati dalle specie reattive dell'ossigeno (ROS) e dagli elettrofili [1]. Le proteine di segnalazione chiave all'interno del percorso sono il fattore di trascrizione Nrf2 (fattore 2 correlato al fattore nucleare eritroide 2) che si lega insieme alle piccole proteine Maf all'elemento di risposta antiossidante (ARE) nelle regioni regolatrici dei geni bersaglio e Keap1 (Kelch ECH associando la proteina 1), una proteina repressiva che si lega a Nrf2 e ne promuove la degradazione da parte della via dell'ubiquitina proteasoma (Fig.1). Keap1 è una proteina molto ricca di cisteina, la Keap1 di topo ha un totale di 25 residui di cisteina umana e 27 di cisteina umana, la maggior parte dei quali può essere modificata in vitro da diversi ossidanti ed elettrofili [2]. Tre di questi residui, C151, C273 e C288, hanno dimostrato di svolgere un ruolo funzionale alterando la conformazione di Keap1 che porta alla traslocazione nucleare di Nrf2 e alla successiva espressione del gene bersaglio [3] (Fig. 1). Il meccanismo esatto per cui le modifiche della cisteina in Keap1 portano all'attivazione di Nrf2 non è noto, ma i due modelli prevalenti ma non si escludono a vicenda sono (1) il modello "cerniera e chiusura", in cui le modifiche di Keap1 nei residui tiolici residenti nell'IVR di Keap1 interrompere l'interazione con Nrf2 causando un disallineamento dei residui di lisina all'interno di Nrf2 che non può più essere polyubiquitinylated e (2) il modello in cui la modifica del tiolo provoca la dissociazione di Cul3 da Keap1 [3]. In entrambi i modelli, il Keap2 modificato dall'induttore e legato a Nrf1 è inattivato e, di conseguenza, le proteine Nrf2 di nuova sintesi bypassano Keap1 e si traslocano nel nucleo, si legano all'ARE e guidano l'espressione dei geni bersaglio Nrf2 come NAD (P) H chinone ossidoreduttasi 1 (NQO1), eme ossigenasi 1 (HMOX1), glutammato-cisteina ligasi (GCL) e glutatione S transferasi (GST) (Fig.2). Oltre alle modifiche dei tioli Keap1 che determinano l'induzione del gene bersaglio Nrf2, proteine come p21 e p62 possono legarsi a Nrf2 o Keap1 interrompendo così l'interazione tra Nrf2 e Keap1 [1], [3] (Fig. 3).
Fig. 1. Strutture di Nrf2 e Keap1 e codice della cisteina. (A) Nrf2 è costituito da 589 amminoacidi e ha sei domini evolutivamente altamente conservati, Neh1-6. Neh1 contiene un motivo bZip, una struttura di base della cerniera leucina (L-Zip), dove la regione di base è responsabile del riconoscimento del DNA e la L-Zip media la dimerizzazione con piccole proteine Maf. Neh6 funziona come un degron per mediare la degradazione di Nrf2 nel nucleo. Neh4 e 5 sono domini di transattivazione. Neh2 contiene motivi ETGE e DLG, necessari per l'interazione con Keap1, e una regione idrofila di residui di lisina (7 K), che sono indispensabili per la poliubiquitinazione dipendente da Keap1 e la degradazione di Nrf2. (B) Keap1 è costituito da 624 residui di amminoacidi e ha cinque domini. I due motivi di interazione proteina-proteina, il dominio BTB e il dominio Kelch, sono separati dalla regione intermedia (IVR). Il dominio BTB insieme alla porzione N-terminale dell'IVR media l'omodimerizzazione di Keap1 e il legame con Cullin3 (Cul3). Il dominio Kelch e la regione C-terminale mediano l'interazione con Neh2. (C) Nrf2 interagisce con due molecole di Keap1 attraverso i suoi motivi Neh2 ETGE e DLG. Sia ETGE che DLG si legano a siti simili sulla superficie inferiore del motivo Keap1 Kelch. (D) Keap1 è ricco di residui di cisteina, con 27 cisteine nelle proteine umane. Alcune di queste cisteine si trovano vicino a residui basici e sono quindi ottimi bersagli di elettrofili e ossidanti. Il modello di modifica dei residui di cisteina da parte degli elettrofili è noto come codice della cisteina. L'ipotesi del codice della cisteina propone che agenti attivanti Nrf2 strutturalmente differenti influenzino diverse cisteine Keap1. Le modifiche della cisteina portano a cambiamenti conformazionali nel Keap1 che interrompono l'interazione tra i domini Nrf2 DLG e Keap1 Kelch, inibendo così la poliubiquitinazione di Nrf2. È stata dimostrata l'importanza funzionale di Cys151, Cys273 e Cys288, poiché Cys273 e Cys288 sono necessari per la soppressione di Nrf2 e Cys151 per l'attivazione di Nrf2 da parte degli induttori [1], [3].
Fig. 2. Il percorso di segnalazione Nrf2-Keap1. (A e B) in condizioni basali, due molecole di Keap1 si legano a Nrf2 e Nrf2 è polyubiquitylated dal complesso di ligasi E3 a base di Cul3. Questa poliubiquitilazione provoca una rapida degradazione di Nrf2 da parte del proteasoma. Una piccola parte di Nrf2 sfugge al complesso inibitorio e si accumula nel nucleo per mediare l'espressione genica basale dipendente da ARE, mantenendo così l'omeostasi cellulare. (C) In condizioni di stress, gli induttori modificano le cisteine Keap1 portando all'inibizione dell'ubiquitylation di Nrf2 attraverso la dissociazione del complesso inibitorio. (D) Secondo il modello di cerniera e latch, la modifica di specifici residui di cisteina Keap1 porta a cambiamenti conformazionali in Keap1 con conseguente distacco del motivo Nrf2 DLG da Keap1. L'ubiquitinazione di Nrf2 è interrotta ma il legame con il motivo ETGE rimane. (E) Nel modello di dissociazione Keap1-Cul3, il legame di Keap1 e Cul3 viene interrotto in risposta agli elettrofili, portando alla fuga di Nrf2 dal sistema di ubiquitinazione. In entrambi i modelli suggeriti, Keap2 legato all'impulso modificato e Nrf1 viene inattivato e, di conseguenza, le nuove proteine Nrf2 sintetizzate bypassano Keap1 e si traslocano nel nucleo, si legano all'elemento di risposta antiossidante (ARE) e guidano l'espressione del bersaglio Nrf2 geni come NQO1, HMOX1, GCL e GST [1], [3].
Fig. 3. Meccanismi per l'accumulo nucleare costitutivo di Nrf2 nel cancro. (A) Le mutazioni somatiche in Nrf2 o Keap1 interrompono l'interazione di queste due proteine. In Nrf2, le mutazioni influenzano i motivi di ETGE e DLG, ma nelle mutazioni di Keap1 sono distribuite in modo più uniforme. Inoltre, l'attivazione di oncogeni, come KrasG12D [5], o l'interruzione di soppressori del tumore, come PTEN [11] può portare all'induzione della trascrizione di Nrf2 e ad un aumento del Nrf2 nucleare. (B) L'ipermetilazione del promotore di Keap1 nel cancro del polmone e della prostata porta alla riduzione dell'espressione di mRNA di Keap1, che aumenta l'accumulo nucleare di Nrf2 [6], [7]. (C) Nel carcinoma papillare renale familiare, la perdita di attività enzimatica dell'idratasi di fumarato porta all'accumulo di fumarato e alla succinazione dei residui di cisteina Keap1 (2SC). Questa modifica post-traduzionale porta all'interruzione dell'interazione di Keap1-Nrf2 e all'accumulo nucleare di Nrf2 [8], [9]. (D) L'accumulo di proteine disgregatrici come p62 e p21 può disturbare il legame Nrf2-Keap1 e determina un aumento del Nrf2 nucleare. p62 si lega a Keap1 sovrapponendosi alla tasca di rilegatura per Nrf2 e p21 interagisce direttamente con i motivi DLG e ETGE di Nrf2, concorrendo così con Keap1 [10].
Meccanismi di attivazione e disregolazione di Nrf2 in Cancro
Sebbene la citoprotezione fornita dall'attivazione di Nrf2 sia importante per la chemioprevenzione del cancro nei tessuti normali e premaligni, nelle cellule completamente maligne l'attività di Nrf2 fornisce un vantaggio di crescita aumentando la chemioresistenza del cancro e migliorando la crescita delle cellule tumorali [4]. Sono stati descritti diversi meccanismi mediante i quali la via di segnalazione Nrf2 è costitutivamente attivata in vari tumori: (1) mutazioni somatiche in Keap1 o nel dominio di associazione di Keap1 di Nrf2 che interrompe la loro interazione; (2) silenziamento epigenetico dell'espressione di Keap1 che porta alla repressione difettosa di Nrf2; (3) accumulo di proteine disgregatrici come p62 che portano alla dissociazione del complesso Keap1-Nrf2; (4) induzione trascrizionale di Nrf2 da oncogenico K-Ras, B-Raf e c-Myc; e (5) modificazione post-traduzionale di cisteine Keap1 da succinilazione che si verifica nel carcinoma renale papillare familiare a causa della perdita di fumarato enzimatica idratasi [3], [4], [5], [6], [7], [ 8], [9], [10] (Fig. 3). La proteina Nrf2 costitutivamente abbondante causa un aumento dell'espressione di geni coinvolti nel metabolismo del farmaco aumentando così la resistenza ai farmaci chemioterapici e alla radioterapia. Inoltre, il livello elevato di proteine Nrf2 è associato a prognosi sfavorevole nel cancro [4]. L'uso eccessivo di Nrf2 influenza anche la proliferazione cellulare indirizzando glucosio e glutammina verso vie anaboliche che aumentano la sintesi della purina e influenzano la via del pentoso fosfato per promuovere la proliferazione cellulare [11] (Fig. 4).
Fig. 4. Il duplice ruolo di Nrf2 nella tumorigenesi. In condizioni fisiologiche, bassi livelli di Nrf2 nucleari sono sufficienti per il mantenimento dell'omeostasi cellulare. Nrf2 inibisce l'iniziazione del tumore e le metastasi del cancro eliminando agenti cancerogeni, ROS e altri agenti dannosi del DNA. Durante la tumorigenesi, l'accumulo di danni al DNA porta a iperattività costitutiva di Nrf2 che aiuta le cellule maligne autonome a sopportare alti livelli di ROS endogena e ad evitare l'apoptosi. Livelli di Nrf2 nucleari persistentemente elevati attivano geni metabolici oltre ai geni citoprotettivi che contribuiscono alla riprogrammazione metabolica e alla proliferazione cellulare potenziata. I tumori con alti livelli di Nrf2 sono associati a prognosi sfavorevole a causa della radio e della chemioresistenza e della proliferazione aggressiva delle cellule tumorali. Pertanto, l'attività del percorso Nrf2 è protettiva nelle prime fasi della tumorigenesi, ma è dannosa nelle fasi successive. Pertanto, per la prevenzione del cancro, l'aumento dell'attività di Nrf2 rimane un approccio importante mentre per il trattamento del cancro è auspicabile l'inibizione di Nrf2 [4], [11].
Dato che l'elevata attività di Nrf2 si verifica comunemente nelle cellule tumorali con esiti avversi, è necessario che le terapie inibiscano Nrf2. Sfortunatamente, a causa della somiglianza strutturale con alcuni altri membri della famiglia bZip, lo sviluppo di specifici inibitori Nrf2 è un compito impegnativo e solo pochi studi sull'inibizione Nrf2 sono stati pubblicati fino ad oggi. Attraverso lo screening di prodotti naturali, Ren et al. [12] ha identificato un composto antineoplastico brusatol come un inibitore Nrf2 che migliora l'efficacia chemioterapica del cisplatino. Inoltre, inibitori di PI3K [11], [13] e Nrf2 siRNA [14] sono stati utilizzati per inibire Nrf2 nelle cellule tumorali. Recentemente, abbiamo utilizzato un approccio alternativo, noto come terapia genica per il suicidio del cancro, per colpire le cellule tumorali con alti livelli di Nrf2. I vettori lentivirali Nrf2-driven [15] contenenti timidina chinasi (TK) sono trasferiti in cellule tumorali con alta attività di ARE e le cellule sono trattate con un pro-farmaco, ganciclovir (GCV). Il GCV viene metabolizzato a GCV-monofosfato, che viene ulteriormente fosforilato dalle chinasi cellulari in una forma trifosfata tossica [16] (Fig. 5). Questo porta ad un'uccisione efficace non solo di cellule tumorali contenenti TK, ma anche delle cellule vicine a causa dell'effetto bystander [17]. La terapia genica TK / GCV regolata con ARE può essere ulteriormente migliorata attraverso la combinazione di un agente chemioterapico chemioterapico doxorubicina nel trattamento [16], supportando la nozione che questo approccio potrebbe essere utile in combinazione con terapie tradizionali.
Fig. 5. Terapia genica suicida. L'accumulo nucleare costitutivo di Nrf2 nelle cellule tumorali può essere sfruttato utilizzando il vettore virale basato su Nrf2 per la terapia genica sul suicidio del cancro [16]. In questo approccio, il vettore lentivirale (LV) che esprime la timidina chinasi (TK) sotto il minimo promotore di SV40 con quattro ARE viene trasdotto in cellule di adenocarcinoma polmonare. Elevati livelli di Nrf2 nucleari portano ad una robusta espressione di TK attraverso il legame Nrf2. Le cellule vengono quindi trattate con un pro-farmaco, ganciclovir (GCV), che viene fosforilato da TK. Il GCV trifosforato altera la sintesi del DNA e porta a un'uccisione efficace non solo delle cellule tumorali contenenti TK, ma anche delle cellule vicine a causa dell'effetto bystander.
Nrf2 è un regolatore principale che attiva la produzione di potenti antiossidanti nel corpo umano che aiutano a eliminare lo stress ossidativo. Diversi enzimi antiossidanti, come la superossido dismutasi, o SOD, glutatione e catalasi, sono attivati anche attraverso il percorso Nrf2. Inoltre, alcune sostanze fitochimiche come la curcuma, l'ashwagandha, la bacopa, il tè verde e il cardo mariano attivano Nrf2. Studi di ricerca lo hanno trovato Attivazione Nrf2 può naturalmente migliorare la protezione cellulare e ripristinare l'equilibrio nel corpo umano.
Dr. Alex Jimenez DC, CCST Insight
Sulforafano e suoi effetti su cancro, mortalità, invecchiamento, cervello e comportamento, malattie cardiache e altro
Gli isotiocianati sono alcuni dei composti vegetali più importanti che si possono ottenere nella dieta. In questo video faccio il caso più completo per loro che sia mai stato fatto. Soglia di attenzione breve? Passa al tuo argomento preferito facendo clic su uno dei seguenti punti temporali. Timeline completa di seguito.
Sezioni chiave:
00: 01: 14 - Cancro e mortalità
00: 19: 04 - Invecchiamento
00: 26: 30 - Cervello e comportamento
00: 38: 06 - Riassunto finale
00: 40: 27 - Dose
Timeline completa:
00: 00: 34 - Introduzione di sulforaphane, uno degli obiettivi principali del video.
00: 01: 14 - Consumo di verdure crocifere e riduzione della mortalità per tutte le cause.
00: 02: 12 - Rischio di cancro alla prostata.
00: 02: 23 - Rischio di cancro alla vescica.
00: 02: 34 - Carcinoma polmonare nei fumatori.
00: 02: 48 - Rischio di cancro al seno.
00: 03: 13 - Ipotetico: cosa succede se hai già un cancro? (Interventistica)
00: 03: 35 - Meccanismo plausibile che guida i dati associativi sul cancro e sulla mortalità.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Prova animale che mostra un forte effetto dell'estratto di germogli di broccolo sullo sviluppo del tumore della vescica nei ratti.
00: 06: 06 - Effetto dell'integrazione diretta di sulforafano nei pazienti affetti da cancro alla prostata.
00: 07: 09 - Bioaccumulo di metaboliti di isotiocianato nel tessuto mammario effettivo.
00: 08: 32 - Inibizione delle cellule staminali del carcinoma mammario.
00: 08: 53 - Lezione di storia: le brassiche sono state istituite come aventi proprietà sanitarie anche nell'antica Roma.
00: 09: 16 - La capacità del Sulforaphane di potenziare l'escrezione di cancerogeno (benzene, acroleina).
00: 09: 51 - NRF2 come interruttore genetico tramite elementi di risposta antiossidante.
00: 10: 10 - Come l'attivazione di NRF2 migliora l'escrezione di cancerogeno tramite glutatione-S-coniugati.
00: 10: 34 - I cavoletti di Bruxelles aumentano la glutatione-S-transferasi e riducono il danno al DNA.
00: 11: 20 - La bevanda di germogli di broccoli aumenta l'escrezione di benzene di 61%.
00: 13: 31 - L'omogenato di germogli di broccoli aumenta gli enzimi antiossidanti nelle vie aeree superiori.
00: 15: 45 - Consumo di verdure crocifere e mortalità per malattie cardiache.
00: 16: 55 - La polvere di germogli di broccoli migliora i lipidi nel sangue e il rischio complessivo di malattie cardiache nei diabetici di tipo 2.
00: 19: 04 - Inizio della sezione di invecchiamento.
00: 19: 21 - La dieta arricchita con sulforafano migliora la durata della vita dei coleotteri da 15 a 30% (in determinate condizioni).
00: 20: 34 - L'importanza della bassa infiammazione per la longevità.
00: 22: 05 - Le verdure crocifere e la polvere di germogli di broccoli sembrano ridurre un'ampia varietà di marcatori infiammatori negli esseri umani.
00: 23: 40 - Ricapitolazione di metà video: cancro, sezioni di invecchiamento
00: 24: 14 - Gli studi sui topi suggeriscono che il sulforafano potrebbe migliorare la funzione immunitaria adattativa in età avanzata.
00: 25: 18 - Sulforaphane ha migliorato la crescita dei peli in un modello murino di calvizie. Immagine su 00: 26: 10.
00: 26: 30 - Inizio della sezione cervello e comportamento.
00: 27: 18 - Effetto dell'estratto di germogli di broccoli sull'autismo.
00: 27: 48 - Effetto del glucorapanin sulla schizofrenia.
00: 28: 17 - Inizio della discussione sulla depressione (meccanismo e studi plausibili).
00: 31: 21 - Lo studio del mouse utilizzando 10 diversi modelli di depressione indotta da stress mostra sulforapano in modo simile efficace come fluoxetina (prozac).
00: 32: 00 - Lo studio mostra che l'ingestione diretta di glucorafanina nei topi è altrettanto efficace nel prevenire la depressione dal modello di stress sociale di sconfitta.
00: 33: 01 - Inizio della sezione di neurodegenerazione.
00: 33: 30 - Sulforaphane e malattia di Alzheimer.
00: 33: 44 - Sulforaphane e morbo di Parkinson.
00: 33: 51 - Sulforaphane e la malattia di Hungtington.
00: 34: 43 - Inizio della sezione traumatica di lesioni cerebrali.
00: 35: 01 - Sulforaphane iniettato immediatamente dopo TBI migliora la memoria (studio del mouse).
00: 35: 55 - Sulforaphane e plasticità neuronale.
00: 36: 32 - Sulforaphane migliora l'apprendimento nel modello di diabete di tipo II nei topi.
00: 37: 19 - Distrofia muscolare sulforapano e duchenne.
00: 37: 44 - Inibizione della miostatina nelle cellule muscolari satelliti (in vitro).
00: 38: 06 - Ricapitolazione tardiva: mortalità e cancro, danno al DNA, stress ossidativo e infiammazione, escrezione di benzene, malattie cardiovascolari, diabete di tipo II, effetti sul cervello (depressione, autismo, schizofrenia, neurodegenerazione), via NRF2.
00: 40: 27 - Pensieri sulla determinazione di una dose di germogli di broccoli o sulforafano.
00: 41: 01 - Aneddoti su germinazione a casa.
00: 43: 14 - Sulle temperature di cottura e sull'attività del sulforafano.
00: 43: 45 - Conversione batterica intestinale del sulforafano da glucorafanina.
00: 44: 24 - Gli integratori funzionano meglio se combinati con la mirosinasi attiva delle verdure.
00: 44: 56 - Tecniche di cottura e verdure crucifere.
00: 46: 06 - Isotiocianati come goitrogens.
Ringraziamenti
Questo lavoro è stato sostenuto dall'Accademia di Finlandia, dalla Sigrid Juselius Foundation e dalle organizzazioni finlandesi per il cancro.
In conclusione, il fattore nucleare (derivato dall'eritroide 2) -come 2, noto anche come NFE2L2 o Nrf2, è una proteina che aumenta la produzione di antiossidanti che proteggono il corpo umano dallo stress ossidativo. Come descritto sopra, la stimolazione del percorso Nrf2 sono in fase di studio per il trattamento di malattie causate da stress ossidativo, compreso il cancro. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
Discussione aggiuntiva sull'argomento: alleviare il dolore al ginocchio senza chirurgia
Il dolore al ginocchio è un sintomo ben noto che può verificarsi a causa di una varietà di lesioni al ginocchio e / o condizioni, tra cui lesioni sportive. Il ginocchio è una delle articolazioni più complesse del corpo umano in quanto è costituito dall'intersezione di quattro ossa, quattro legamenti, vari tendini, due menischi e cartilagine. Secondo l'American Academy of Family Physicians, le cause più comuni di dolore al ginocchio sono la sublussazione patellare, la tendinite rotulea o il ginocchio del saltatore e la malattia di Osgood-Schlatter. Anche se il dolore al ginocchio è più probabile che si verifichi nelle persone sopra 60 anni, il dolore al ginocchio può verificarsi anche nei bambini e negli adolescenti. Il dolore al ginocchio può essere trattato a casa seguendo i metodi del RISO, tuttavia, gravi lesioni al ginocchio possono richiedere cure mediche immediate, inclusa la cura chiropratica.
Il DNA supporta approssimativamente i geni 20,000, ognuno dei quali possiede un programma per la creazione di una proteina o di un enzima necessario per uno stile di vita sano. Ognuno di questi modelli deve essere costantemente regolato da una sorta di "promotore" che gestisce esattamente quanto di ogni sostanza e / o sostanza chimica è generata e in quali condizioni si svilupperanno.
Collegandosi a un particolare tipo di aree del promotore simili a interruttori, noto come l'elemento di risposta antiossidante, o ARE, il Fattore Nrf2Supporta la velocità di creazione di centinaia di geni distinti che consentono alle cellule di sopravvivere in circostanze stressanti. Questi geni generano quindi una selezione di enzimi antiossidanti che sviluppano una rete di difesa neutralizzando gli ossidanti e ripulendo i sottoprodotti tossici lasciati nella loro produzione, oltre ad aiutare a ripristinare i danni che hanno causato.
Cos'è lo stress ossidativo?
Diversi ossidanti come il radicale superossido, o O2-., E il perossido di idrogeno, o H2O2, sono stati creati attraverso la pratica di bruciare le sostanze e / o le sostanze chimiche che sostengono il corpo umano. Il corpo umano possiede enzimi antiossidanti che neutralizzano e disintossicano i cibi e le bevande reattive che consumiamo. Il Nrf2 modula la loro produzione per mantenere l'equilibrio e sottolinea la richiesta di tutti questi enzimi. Questo equilibrio può essere interrotto da una coppia di fattori, inclusa l'età.
Con l'avanzare dell'età, il corpo umano crea meno Nrf2 e questo delicato equilibrio può gradualmente iniziare a girare verso il lato ossidativo, uno stato denominato stress ossidativo. La malattia può anche causare la sovrapproduzione di ossidanti. Infezioni, allergie e disturbi autoimmuni possono inoltre indurre le nostre cellule immunitarie a creare ossidanti reattivi, come l'O2-. , H2O2, OH e HOCl, dove le cellule sane vengono danneggiate e rispondono con l'infiammazione. Anche le malattie associate all'invecchiamento, inclusi attacchi di cuore, ictus, cancro e condizioni neurodegenerative come il morbo di Alzheimer, aumentano lo sviluppo di ossidanti, generando stress e una risposta infiammatoria.
Cosa sono gli attivatori Nrf2?
La proteina Nrf2, chiamata anche fattore di trascrizione per il modo in cui può supportare e controllare enzimi e geni, è l'elemento segreto di una sequenza di reazioni biochimiche all'interno della cellula che reagisce alle modificazioni dell'equilibrio cognitivo e dell'equilibrio ossidativo. Gli elementi sensibili di questo percorso modificano e scaricano Nrf2, attivandolo in modo che possa diffondersi nel nucleo della cellula verso il DNA. Il Nrf2 può alternativamente accendere o spegnere i geni e gli enzimi che supporta per proteggere la cellula.
Fortunatamente, una varietà di sostanze che sono attivatori Nrf2 si sviluppano attraverso il consumo di alcune piante ed estratti utilizzati secoli fa in rimedi tradizionali cinesi e nativi americani. Queste sostanze fitochimiche sembrano altrettanto potenti con minori effetti collaterali, come i prodotti farmaceutici che attivano Nrf2 oggi utilizzati.
Fattore di fattore eritroide Il fattore correlato a 2, più comunemente noto come Nrf2, è un fattore di trascrizione che protegge la cellula regolando i geni, gli enzimi e le risposte antiossidanti. I fattori di trascrizione sono un tipo di proteina che si lega al DNA per promuovere la creazione di sostanze e sostanze chimiche specifiche, tra cui le S-transferasi del glutatione o GST. L'attivazione di Nrf2 induce la produzione di proteine attive che esibiscono una potente capacità antiossidante per aiutare a ridurre lo stress ossidativo.
Dr. Alex Jimenez DC, CCST Insight
La scienza dietro l'attivazione Nrf2
Una volta che il primo integratore alimentare attivante Nrf2 è stato creato in 2004, sono state fornite informazioni minime riguardanti la funzione del percorso Nrf2. Circa i giornali 200 in letteratura su Nrf2, noto anche come 2 a fattore nucleare o NFE2L2, esistevano e i ricercatori stavano appena iniziando a scoprire la risposta antiossidante di Nrf2 nei mammiferi. A partire da 2017, tuttavia, sono stati stampati gli studi di ricerca accademica 9,300 su questo "regolatore principale".
In realtà, Nrf2 regola molti enzimi antiossidanti che non sono correlati ai geni, invece, offrono protezione contro una varietà di circostanze legate allo stress che si verificano in cellule, organi e infine organismi, in condizioni salutari e patologiche. Sulla base di questa nuova quantità di informazioni provenienti da studi di ricerca accademici pubblicati, i ricercatori ora possono svilupparsi meglio Nrf2 integratori alimentari.
A partire dal 2007, studi di ricerca hanno dimostrato la complessa funzione del percorso Nrf2. È stato scoperto che gli attivatori Nrf2 imitano fattori di diverse strutture all'interno del corpo umano. Attraverso questi percorsi, gli attivatori Nrf2 sono stati attrezzati per sentire le condizioni mutevoli in tutta la cellula al fine di mantenere l'equilibrio e rispondere alle esigenze in evoluzione dei geni.
Perché utilizzare i supplementi attivanti Nrf2?
Dato che le capacità di attivazione di Nrf2 diminuiscono con l'età negli organismi, i cambiamenti possono iniziare a verificarsi. Studi di ricerca hanno dimostrato che l'attenzione di Nrf2 nelle cellule diminuisce con l'età, mostrando marcatori aumentati di stress ossidativo. Una varietà di malattie legate all'età come l'aterosclerosi e le malattie cardiovascolari, l'artrite, il cancro, l'obesità, il tipo di diabete 2, l'ipertensione, la cataratta e il morbo di Alzheimer così come le malattie del Parkinson possono svilupparsi a causa di questi cambiamenti. Lo stress ossidativo è stato trovato con questi problemi di salute.
Stimolando la capacità della cellula di aumentare la produzione di attivatori Nrf2, Nrf2 integratori alimentari può aiutare a far rivivere la capacità del corpo umano di contrastare gli effetti dello stress ossidativo. Gli acidi grassi polinsaturi, o PUFA, sono una delle molecole più facilmente ossidabili e sono particolarmente vulnerabili a subire danni da radicali liberi. L'acido tiobarbiturico o TBARS, la produzione può aumentare con l'età, indicando un aumento dello stress ossidativo e una diminuzione dei percorsi regolati da Nrf2.
Biologicamente, l'induzione genica è un meccanismo molto lento, che generalmente richiede ore per il trasferimento attraverso un percorso. Di conseguenza, molti enzimi possiedono i propri interruttori di accensione / spegnimento che potrebbero essere attivati in pochi minuti da diversi enzimi regolatori. I ricercatori hanno sviluppato composizioni proprietarie di attivatori Nrf2 che utilizzano questa base di conoscenza dell'attivazione. L'attivazione di Nrf2 è composta non solo dal fattore di trascrizione Nrf2 che viene scaricato dal suo inibitore e che migra al nucleo cellulare, ma si lega anche a specifiche sequenze di DNA per incoraggiare l'espressione genica citoprotettiva, regolando il ritmo con cui Nrf2 viene estratto dal nucleo.
Comprendere la procedura di eliminazione e l'attivazione di Nrf2 nel corpo umano ha permesso ai ricercatori di costruire combinazioni di diversi attivatori Nrf2 per realizzare la riflessione dei geni attraverso la sua modulazione. La combinazione della base di conoscenze, insieme all'ampia varietà di altri studi di ricerca, ha contribuito a produrre attivatori Nrf2 da utilizzare come integratori alimentari. Lo scopo delle nostre informazioni è limitato ai problemi di salute della colonna vertebrale e della chiropratica. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
A cura di Dr. Alex Jimenez
Discussione aggiuntiva sull'argomento: alleviare il dolore al ginocchio senza chirurgia
Il dolore al ginocchio è un sintomo ben noto che può verificarsi a causa di una varietà di lesioni al ginocchio e / o condizioni, tra cui lesioni sportive. Il ginocchio è una delle articolazioni più complesse del corpo umano in quanto è costituito dall'intersezione di quattro ossa, quattro legamenti, vari tendini, due menischi e cartilagine. Secondo l'American Academy of Family Physicians, le cause più comuni di dolore al ginocchio sono la sublussazione patellare, la tendinite rotulea o il ginocchio del saltatore e la malattia di Osgood-Schlatter. Anche se il dolore al ginocchio è più probabile che si verifichi nelle persone sopra 60 anni, il dolore al ginocchio può verificarsi anche nei bambini e negli adolescenti. Il dolore al ginocchio può essere trattato a casa seguendo i metodi del RISO, tuttavia, gravi lesioni al ginocchio possono richiedere cure mediche immediate, inclusa la cura chiropratica.
Gli antiossidanti sono definiti scientificamente come composti che limitano il processo di ossidazione nel corpo umano, che se non controllati, possono creare radicali liberi che possono sviluppare numerose reazioni a catena che possono causare danni cellulari. Fortunatamente, il corpo umano può creare tali meccanismi immunitari incorporati, tuttavia, quando le specie reattive dell'ossigeno, o ROS, non sono in grado di essere neutralizzate, immaginano una minuscola fiamma che diventa fuori controllo quando infusa con ossigeno, il danno è destinato a verificarsi .
Per continuare ad espandersi sulla metafora della fiamma, il prodotto finale di non avere la capacità di neutralizzare l'impatto delle ROS, o specie reattive dell'ossigeno, è il danno e l'infiammazione, in altre parole, il corpo umano è letteralmente in fiamme. La cosa fantastica è che ci sono antiossidanti che possono tremendamente aiutare a combattere questo problema di salute e questo antiossidante è il glutatione. Sebbene trovato in 1889, l'effetto antiossidante del glutatione è diventato uno degli argomenti più interessanti negli studi di ricerca moderni.
Maestro di antiossidanti: glutatione
La potente sostanza è un tripeptide che si sviluppa da cisteina, acido glutammico e glicina. A causa della sua capacità di proteggere il corpo umano dalla creazione di radicali liberi, il glutatione può aiutare a promuovere un sistema immunitario sano. Basato su Rapporti scientifici in 2015, è stato determinato che la capacità del glutatione di funzionare sinergicamente con peroxiredina e catalasi aiuta a proteggere le cellule dal perossido di idrogeno. Questa formula sinergica funziona contro le specie reattive dell'ossigeno, o ROS. Il glutatione, la perossididina e la catalasi sono elementi essenziali nell'aumento dell'omeostasi cellulare, che è un processo essenziale di cellule, tessuti e organi sani.
Inoltre, il glutatione aumenta la struttura e la funzione del sistema immunitario generale, utilizzando il suo importante effetto sulle funzioni dei linfociti. Secondo il Dipartimento di Immunochimica, integrando correttamente i livelli di glutatione nel corpo umano può migliorare notevolmente le reazioni immunitarie. A titolo di esempio, due studi randomizzati controllati verso placebo hanno dimostrato che il trattamento terapeutico di pazienti immunocompromessi con N-acetil-cisteina, o NAC, risultava, in entrambi i casi, in una crescita sostanziale nella maggior parte dei processi immunologici che includevano un intero ringiovanimento dell'attività naturale delle cellule killer. N-acetil-cisteina, o NAC, utilizza lo zolfo del glutatione e lo combina con molecole velenose, che quindi diventano solubili in acqua e vengono scaricate nel corpo umano.
Il glutatione ha anche la capacità di rivitalizzare l'acido lipoico e di riciclare la vitamina C ed E, che sono necessarie per avviare determinati processi del sistema inviando elettroni per neutralizzare i radicali liberi. Basato su uno studio di ricerca di PLoS ONE, pazienti affetti da glutatione con diabete metillus, o T2DM e micobatterio tubercolosi. Normalmente, gli individui con un sistema immunitario debole hanno la tendenza a mostrare una maggiore esposizione a M. tb, o micobatterio tubercolosi, malattia o infezione. Inoltre, le persone con diabete di tipo 2 metilllus, o T2DM, sono da due a tre volte più soggette a TB rispetto alle persone senza T2DM. Lo studio ha anche suggerito che l'aumento dei livelli di glutatione nei macrofagi isolati da pazienti con T2DM ha portato a un migliore controllo della malattia o infezione da M.Tb. Questi risultati dimostrano che livelli più bassi di glutatione nei pazienti con T2DM contribuisce ad aumentare la probabilità di malattia o infezione da M. tb. Inoltre, dipende da Dietro Ghezzi a Brighton e Sussex Medical Schoollo stress ossidativo può infine causare una cattiva struttura e funzione del sistema immunitario.
Fortunatamente, il glutatione svolge un ruolo essenziale nel rafforzare e controllare l'immunità. A titolo di esempio, il glutatione è essenziale per i processi innati e adattivi all'interno del sistema immunitario, compresa la proliferazione dei linfociti T, l'attività fagocitaria dei neutrofili polimorfonucleati e le funzioni delle cellule dendritiche, che possono essere fondamentali perché sono costituite da cellule che presentano l'antigene . L'immunità meditata dalle cellule include antigeni proteici che inizialmente iniziano a degenerare nelle vescicole endocitiche di macrofagi e cellule dendritiche, pertanto i peptidi più piccoli sono dimostrati sulla superficie per attivare la proliferazione di cellule T antigene-specifiche. Inoltre, il glutatione contribuisce alla creazione di citochine ed è necessario mantenere la produzione di interferone-gamma da parte delle cellule dendritiche, che è importante per proteggere contro i patogeni intracellulari, compresi i micobatteri.
N-acetil-cisteina, o NAC, scientificamente indicato come il precursore del glutatione, è anche un potente antiossidante cellulare usato come antiossidante scavenger di radicali liberi. Comunemente riconosciuto per il suo ruolo in evitando la tossicità del paracetamolo, NAC, o N-acetil-cisteina, ha dimostrato di possedere numerosi benefici per la salute e il benessere. Secondo Diario delle cellule, NAC aiuta a sostenere una risposta infiammatoria sana e può avere un impatto positivo sul termine umano e sui lavori pretermine. Lo studio ha concluso che nelle donne con pregressa nascita pretermine e vaginosi batterica, il glicemia 0.6 al giorno assunta per via orale insieme al progesterone dopo la settimana 16 della gravidanza ha protetto la recidiva di nascita pretermine e ha migliorato i risultati neonatali. In conclusione, sono stati rilevati anche gli effetti positivi del NAC sulla costruzione muscolare. Dopo tre minuti di contrazioni persistenti, c'è stato un aumento della percentuale di 15, a dimostrazione di come NAC svolge un ruolo fondamentale nel miglioramento della costruzione muscolare e nella riduzione dell'affaticamento generale durante il travaglio.
I ricercatori hanno anche scoperto che la NAC, o N-acetil-cisteina, può giovare a coloro che hanno la sindrome dell'ovaio policistico o PCOS. La PCOS, o sindrome dell'ovaio policistico, è una comune malattia correlata alle ghiandole endocrine che colpisce circa il 5-10% delle donne in età riproduttiva. In questi pazienti, c'è un rischio maggiore di sperimentare la sindrome metabolica, in cui l'uso di NAC ha contribuito a ripristinare livelli sani di insulina e sensibilità.
Insight di Dr. Alex Jimenez
Il glutatione è stato definito il "maestro degli antiossidanti" per il suo ruolo fondamentale nel raggiungimento e nel mantenimento della salute e del benessere generale. Mentre il corpo umano è in grado di produrre il proprio glutatione, cattiva alimentazione, inquinamento, tossine, uso eccessivo di droghe e / o farmaci, stress, traumi, invecchiamento, malattie e radiazioni possono tutti diminuire i nostri livelli naturali di glutatione. Ciò può a sua volta rendere gli individui più suscettibili al danno cellulare dallo stress ossidativo, dai radicali liberi, dalle infezioni e dal cancro. L'integrazione di glutatione può quindi avere enormi benefici sul corpo umano. Insieme alle opzioni di trattamento alternative, come la cura chiropratica, i livelli di glutatione possono essere nuovamente regolati per migliorare il benessere.
Inoltre, gli operatori sanitari hanno suggerito di implementare l'uso della supplementazione di glutatione insieme ad altre opzioni di trattamento alternative, come ad esempio cure chiropratiche, per migliorare ulteriormente la salute e il benessere generale. Gli antiossidanti sono importanti per il mantenimento del massimo benessere e per inibire la reazione a catena dei radicali liberi che causano danni o danni alle cellule. Potenti antiossidanti come il glutatione, come accennato in precedenza, in definitiva aiutano a regolare lo sviluppo di questi radicali liberi e forniscono una risposta del sistema immunitario più sana. Studi di ricerca lo hanno trovato cure chiropratiche può anche svolgere un ruolo essenziale in questo processo, aumentando naturalmente l'attività degli antiossidanti nel corpo umano. La cura chiropratica è un approccio terapeutico sicuro ed efficace che utilizza regolazioni spinali e manipolazioni manuali per correggere i disallineamenti spinali, o sublussazioni, al fine di consentire al corpo umano di guarire naturalmente se stesso senza l'uso di farmaci / farmaci e / o interventi chirurgici.
Infine, gli antiossidanti dimostrano le loro proprietà biologiche attraverso una grande quantità di benefici per la salute, che potrebbero essere necessari ora più che mai con ogni crescente assalto di stress, malattie e inquinamento nel nostro mondo moderno, che contribuiscono tutti al danno e / o al danno cellulare . Il glutatione e il suo precursore, NAC, o N-acetil-cisteina, continuano a mostrare il loro potente status nel regno degli antiossidanti. Insieme a opzioni di trattamento alternative, come la cura chiropratica, le persone possono trarre vantaggio da tutti i benefici che questo potente antiossidante ha da offrire. Lo scopo delle nostre informazioni è limitato alla chiropratica e alle lesioni e condizioni della colonna vertebrale. Per discutere l'argomento, non esitate a chiedere al Dr. Jimenez o contattarci a 915-850-0900 .
A cura di Dr. Alex Jimenez
Argomenti aggiuntivi: Dolore alla schiena
Mal di schiena è una delle cause prevalenti di disabilità e giornate perse al lavoro in tutto il mondo. Di fatto, il dolore alla schiena è stato attribuito come la seconda ragione più comune per le visite di un medico, superata solo dalle infezioni delle alte vie respiratorie. Circa il 80 percento della popolazione sperimenterà qualche tipo di dolore alla schiena almeno una volta nel corso della vita. La colonna vertebrale è una struttura complessa composta da ossa, articolazioni, legamenti e muscoli, tra gli altri tessuti molli. A causa di ciò, lesioni e / o condizioni aggravate, come dischi erniciati, può eventualmente portare a sintomi di mal di schiena. Le lesioni sportive o gli incidenti automobilistici sono spesso la causa più frequente di mal di schiena, tuttavia a volte il più semplice dei movimenti può avere risultati dolorosi. Fortunatamente, le opzioni di trattamento alternative, come la cura chiropratica, possono aiutare ad alleviare il mal di schiena attraverso l'uso di aggiustamenti spinali e manipolazioni manuali, in definitiva migliorando il sollievo dal dolore.
Chiropratico basato sulla scienza Dr. Alexander Jimenez dà un'occhiata lo stress ossidativo, che cosa è, come colpisce il corpo e la difesa antiossidante per rimediare alla situazione.
Esra Birben PhD, 1 Umit Murat Sahiner MD, 1 Cansin Sackesen MD, 1 Serpil Erzurum MD, 2 e Omer Kalayci, MD1
Abstract: Le specie reattive dell'ossigeno (ROS) sono prodotte da organismi viventi come risultato del normale metabolismo cellulare e di fattori ambientali, come gli inquinanti atmosferici o il fumo di sigaretta. I ROS sono molecole altamente reattive e possono danneggiare le strutture cellulari come carboidrati, acidi nucleici, lipidi e proteine e alterarne le funzioni. Lo spostamento dell'equilibrio tra ossidanti e antiossidanti a favore degli ossidanti è definito `` stress ossidativo ''. La regolazione dello stato riducente e ossidante (redox) è fondamentale per la vitalità, l'attivazione, la proliferazione e la funzione degli organi delle cellule. Gli organismi aerobici hanno sistemi antiossidanti integrati, che includono antiossidanti enzimatici e non enzimatici che di solito sono efficaci nel bloccare gli effetti nocivi dei ROS. Tuttavia, in condizioni patologiche, i sistemi antiossidanti possono essere sopraffatti. Lo stress ossidativo contribuisce a molte condizioni patologiche e malattie, tra cui cancro, disturbi neurologici, aterosclerosi, ipertensione, ischemia / perfusione, diabete, sindrome da distress respiratorio acuto, fibrosi polmonare idiopatica, broncopneumopatia cronica ostruttiva e asma. In questa recensione, riassumiamo i sistemi ossidanti e antiossidanti cellulari e discutiamo gli effetti cellulari ei meccanismi dello stress ossidativo.
Parole chiave: antiossidante, ossidante, stress ossidativo, specie di ossigeno reattivo, redox
(WAO Journal 2012; 5: 9-19)
Le specie reattive dell'ossigeno (ROS) sono prodotte da organismi viventi come risultato del normale metabolismo cellulare. A concentrazioni da basse a moderate, funzionano nei processi cellulari fisiologici, ma ad alte concentrazioni producono modifiche negative ai componenti cellulari, come lipidi, proteine e DNA.1 6 Lo spostamento dell'equilibrio tra ossidante / antiossidante a favore degli ossidanti è definito `` stress ossidativo ''. Lo stress ossidativo contribuisce a molte condizioni patologiche, tra cui cancro, disturbi neurologici, aterosclerosi 7-10, ipertensione, ischemia / perfusione, diabete 11-14, sindrome da distress respiratorio acuto, fibrosi polmonare idiopatica, malattia polmonare ostruttiva cronica , 15 e l'asma.16 21 Gli organismi aerobici hanno sistemi antiossidanti integrati, che includono antiossidanti enzimatici e non enzimatici che di solito sono efficaci nel bloccare gli effetti nocivi dei ROS. Tuttavia, in condizioni patologiche, i sistemi antiossidanti possono essere sopraffatti. In questa recensione, riassumiamo i sistemi ossidanti e antiossidanti cellulari e la regolazione dello stato riducente e ossidante (redox) negli stati di salute e malattia.
OSSIDANTI
Fonti endogene di ROS
I ROS sono prodotti da ossigeno molecolare come risultato di un normale metabolismo cellulare. ROS può essere diviso in gruppi 2: radicali liberi e nonradicali. Molecole contenenti uno o più elettroni sconosciuti e dando così reattività alla molecola sono chiamati radicali liberi. Quando i radicali liberi di 2 condividono i loro elettroni non accoppiati, vengono create forme nonradiche. I principali ROS 3, che sono di significato fisiologico, sono l'anione superossido (O22.), Il radicale idrossile (OH) e il perossido di idrogeno (H2O2). ROS sono riassunti nella Tabella 1.
L'anione superossido è formato dall'aggiunta di 1 elettrone all'ossigeno molecolare.22 Questo processo è mediato dalla nicotina adenina dinucleotide fosfato [NAD (P) H] ossidasi o xantina ossidasi o dal sistema di trasporto di elettroni mitocondriali. Il sito principale per la produzione di anione superossido sono i mitocondri, il meccanismo della cellula per produrre adenosina trifosfato. Normalmente, gli elettroni vengono trasferiti attraverso la catena di trasporto degli elettroni mitocondriale per la riduzione dell'ossigeno all'acqua, ma circa l'1-3% di tutti gli elettroni fuoriesce dal sistema e produce superossido. La NAD (P) H ossidasi si trova nei leucociti polimorfonucleati, nei monociti e nei macrofagi. Dopo la fagocitosi, queste cellule producono un'esplosione di superossido che porta all'attività battericida. Il superossido viene convertito in perossido di idrogeno dall'azione delle superossido dismutasi (SOD, EC 1.15.1.1). Il perossido di idrogeno si diffonde facilmente attraverso la membrana plasmatica. Il perossido di idrogeno è anche prodotto dalla xantina ossidasi, dall'amminoacido ossidasi e dalla NAD (P) H ossidasi 23,24 e nei perossisomi dal consumo di ossigeno molecolare nelle reazioni metaboliche. In una successione di reazioni chiamate reazioni di Haber Weiss e Fenton, H2O2 può degradarsi in OH2 in presenza di metalli di trasmissione come Fe21 o Cu21.25
Fe31 + .O2 ? Fe2 + O2 Haber Weiss
Fe2 + H2O2 ? Fe3 + OH + .OH Reazione di Fenton
L'O 2 stesso può anche reagire con H2 O2 e generare OH.26,27 Il radicale idrossile è il più reattivo dei ROS e può danneggiare proteine, lipidi, carboidrati e DNA. Può anche iniziare la perossidazione lipidica prendendo un elettrone dagli acidi grassi polinsaturi.
Gli enzimi granulocitici espandono ulteriormente la reattività di H2O2 tramite la perossidasi eosinofila e la mieloperossidasi (MPO). Nei neutrofili attivati, H2O2 viene consumato da MPO. In presenza di ione cloruro, H2O2 viene convertito in acido ipocloroso (HOCl). HOCl è altamente ossidante e svolge un ruolo importante nell'uccisione dei patogeni nelle vie aeree.28 Tuttavia, HOCl può anche reagire con il DNA e indurre interazioni DNA proteine e produrre prodotti di ossidazione della pirimidina e aggiungere cloruro alle basi del DNA.29,30 Perossidasi eosinofila e MPO contribuiscono anche allo stress ossidativo modificando le proteine mediante alogenazioni, nitrazione e legami crociati di proteine tramite i radicali tirosilici.
Altri radicali liberi derivati dall'ossigeno sono i radicali perossilici (ROO $). La forma più semplice di questi radicali è il radicalo idroperossilico (HOO $) e ha un ruolo nella perossidazione dell'acido grasso. I radicali liberi possono innescare le reazioni a catena della perossidazione lipidica estraggendo un atomo di idrogeno da un carbonio di metilene a catena laterale. Il radicale lipidico quindi reagisce con l'ossigeno per produrre radicali perossilici. Il radical perossilico inizia una reazione a catena e trasforma gli acidi grassi polinsaturi in idroperossidi lipidi. Gli idroperossidi lipidici sono molto instabili e possono facilmente decomporsi a prodotti secondari, quali aldeidi (come 4-idrossi-2,3-nonenali) e malondialdehidi (MDA). Gli isoprostani sono un altro gruppo di prodotti perossidanti lipidi che vengono generati attraverso la perossidazione dell'acido arachidonico e sono stati trovati anche elevati in plasma e respirazione di condensati degli asmatici.34,35 La perossidazione dei lipidi disturba l'integrità delle membrane cellulari e porta alla riorganizzazione della struttura a membrana .
Perossido di idrogeno, superossido radicale, glutatione ossidato (GSSG), MDAs, isoprostani, carbonili e nitrotirosina possono essere misurati facilmente dai campioni di lavaggio del plasma, del sangue o del broncoalveolare come biomarkers di ossidazione mediante saggi standardizzati.
Origine esogena di ossidanti
Fumo di sigaretta
Il fumo di sigaretta contiene molti ossidanti e radicali liberi e composti organici, come il superossido e l'ossido nitrico. 36 Inoltre, l'inalazione di fumo di sigarette nel polmone attiva anche alcuni meccanismi endogeni, come l'accumulo di neutrofili e macrofagi, che aumentano ulteriormente l'infortunio ossidante .
Esposizione all'ozono
L'esposizione all'ozono può causare la perossidazione dei lipidi e indurre l'afflusso di neutrofili nell'epitelio delle vie aeree. L'esposizione a breve termine all'ozono provoca anche il rilascio di mediatori infiammatori, come MPO, proteine cationiche eosinofili e anche lattato deidrogenasi e albumina. NUMX Anche nei soggetti sani l'esposizione all'ozono provoca una riduzione delle funzioni polmonari.37 Cho et al38 hanno dimostrato che particolato (miscela di particelle solide e goccioline di liquido sospese nell'aria) catalizza la riduzione dell'ossigeno.
iperossia
L'iperossia si riferisce a condizioni di elevati livelli di ossigeno rispetto alla normale pressione parziale di ossigeno nei polmoni o in altri tessuti del corpo. Porta a una maggiore produzione di ossigeno reattivo e specie di azoto
Radiazione ionizzante
La radiazione ionizzante, in presenza di O2, converte il radicale idrossile, il superossido e i radicali organici in perossido di idrogeno e idroperossidi organici. Queste specie di idroperossido reagiscono con gli ioni metallici attivi redox, come Fe e Cu, tramite reazioni di Fenton e quindi inducono stress ossidativo.42,43 Narayanan et al44 hanno mostrato che i fibroblasti che erano stati esposti a particelle alfa avevano aumenti significativi di O2 2. intracellulare e H2O2 produzione tramite NADPH ossidasi legata alla membrana plasmatica.44 Molecole di trasduzione del segnale, come la chinasi 1 e 2 extracellulare regolata dal segnale (ERK1 / 2), la chinasi N-terminale c-Jun (JNK) e p38 e fattori di trascrizione, come la proteina attivatore-1 (AP-1), il fattore nucleare-kB (NF-kB) e la p53 vengono attivati, il che determina l'espressione di geni correlati alla risposta alle radiazioni. per eccitazione di fotosensibilizzatori endogeni, come porfirine, NADPH ossidasi e riboflavine. L'45-Oxo-50- diidroguanina (8-oxoGua) è il principale prodotto di ossidazione del DNA mediato da UVA formato dall'ossidazione del radicale OH, degli ossidanti 7,8-elettrone e dell'ossigeno singoletto che reagisce principalmente con la guanina.8 La formazione di guanina È stato dimostrato che il catione radicalico nel DNA isolato si verifica in modo efficiente attraverso l'effetto diretto delle radiazioni ionizzanti.1 Dopo l'esposizione a radiazioni ionizzanti, il livello intracellulare di glutatione (GSH) diminuisce per un breve periodo ma poi aumenta di nuovo.51
Ioni di metalli pesanti
I ioni di metalli pesanti, quali il ferro, il rame, il cadmio, il mercurio, il nichel, il piombo e l'arsenico, possono indurre la generazione di radicali reattivi e causare danni cellulari attraverso l'esaurimento delle attività enzimatiche attraverso la perossidazione dei lipidi e la reazione con proteine nucleari e DNA.
Uno dei meccanismi più importanti della generazione di radicali liberi mediata da metalli è attraverso una reazione di tipo Fenton. Lo ione superossido e il perossido di idrogeno possono interagire con i metalli di transizione, come il ferro e il rame, tramite la reazione di HaberWeiss / Fenton catalizzata dal metallo per formare radicali OH.
Oltre ai meccanismi di tipo Fenton e Haber-Weiss, alcuni ioni metallici possono reagire direttamente con le molecole cellulari per generare radicali liberi, come i radicali tiolici, o indurre percorsi di segnalazione cellulare. Questi radicali possono anche reagire con altre molecole di tiolo per generare O22 .. O22. viene convertito in H2O2, che provoca la generazione di radicali ossigeno aggiuntivi. Alcuni metalli, come l'arsenite, inducono indirettamente la formazione di ROS mediante l'attivazione di sistemi di produzione di radicali nelle cellule. 56
L'arsenico è un elemento altamente tossico che produce una varietà di ROS, tra cui superossido (O2 2), ossigeno singoletto (1O2), radicale perossilico (ROO), ossido nitrico (NO), perossido di idrogeno (H2O2) e radicali perossilici dimetilarsinici [( CH3) 2AsOO] .57 I composti dell'arsenico (III) possono inibire gli enzimi antiossidanti, in particolare gli enzimi dipendenti dal GSH, come la glutatione-S-transferasi (GST), la glutatione perossidasi (GSH-Px) e la GSH reduttasi, tramite legame - ing ai loro gruppi solfidrilici ( SH) .59
Il piombo aumenta la perossidazione dei lipidi. 62 La riduzione significativa dell'attività del tessuto SOD e del cervello GPx è stata riportata dopo l'esposizione al piombo.63,64 La sostituzione dello zinco, che funge da cofattore per molti enzimi da piombo, porta all'attivazione di tali enzimi. L'esposizione del piombo può causare inibizione del GST colpendo i tioli dei tessuti.
ROS generato da reazioni catalizzate con metallo può modificare le basi del DNA. Possono verificarsi tre sostituzioni di base, G / C, G / T e C / T, in seguito a danni ossidativi da ioni metallici, quali Fe21, Cu21 e Ni21. Reid et al65 ha mostrato che G / C è stato prevalentemente prodotto da Fe21 mentre la sostituzione C / T è stata effettuata da Cu21 e Ni21.
ANTIOSSIDANTI
Il corpo umano è dotato di una varietà di antiossidanti che servono a controbilanciare l'effetto degli ossidanti. Per tutti gli scopi pratici questi possono essere suddivisi in categorie 2: enzimatiche (Tabella 2) e non-enzimatiche (Tabella 3).
Antiossidanti enzimatici
I principali antiossidanti enzimatici dei polmoni sono SOD (EC 1.15.1.11), catalasi (EC 1.11.1.6) e GSH-Px (EC 1.11.1.9). Oltre a questi principali enzimi, sono stati trovati anche altri antiossidanti, tra cui l'emenossigenasi-1 (EC 1.14.99.3) e le proteine redox, quali le tioredossine (TRXs, EC1.8.4.10), perossiredossine (PRXs, EC1.11.1.15) e glutareossine svolgono un ruolo cruciale nelle difese antiossidanti polmonari.
Poiché il superossido è il principale ROS prodotto da una varietà di sorgenti, la sua dissoluzione da SOD è di primaria importanza per ogni cellula. Tutte le forme 3 di SOD, cioè CuZn-SOD, Mn-SOD e EC-SOD, sono ampiamente espresse nel polmone umano. Mn-SOD è localizzato nella matrice mitocondriale. EC-SOD è localizzato principalmente nella matrice extracellulare, specialmente nelle aree contenenti elevate quantità di fibre di collagene di tipo I e intorno a recipienti polmonari e sistemici. È stato anche rilevato nell'epitelio bronchiale, nell'epitelio alveolare e nei macrofagi alveolari. Nel complesso, CuZn-SOD e Mn-SOD sono generalmente pensati di agire come scavenger di massa dei radicali superossidici. Il livello relativamente elevato di EC-SOD nel polmone con il suo legame specifico alle componenti della matrice extracellulare può rappresentare una componente fondamentale della protezione della matrice polmonare.66,67
H2O2 prodotta dall'azione di SOD o dall'azione di ossidasi, come la xantina ossidasi, viene ridotta all'acqua con catalasi e GSH-Px. Catalase esiste come un tetramero composto da monomeri identici 4, ognuno dei quali contiene un gruppo heme nel sito attivo. La degradazione di H2O2 viene effettuata mediante la conversione tra conformazioni 2 della catalasi-ferricatalasi (ferro coordinato all'acqua) e del composto I (ferro complessato con un atomo di ossigeno). La catalasi lega anche NADPH come equivalente riducente per impedire l'inattivazione ossidativa dell'enzima (formazione del composto II) da H2O2 in quanto viene ridotta all'acqua.69
Gli enzimi nel ciclo redox responsabile della riduzione di H2O2 e degli idroperossidi lipidi (generati come risultato di perossidazione lipidica a membrana) includono GSH-Px. 70 Gli GSH-Pxs sono una famiglia di enzimi tetramerici che contengono l'unica selenocisteina dell'amminoacido all'interno del siti attivi e utilizzare tioli a basso peso molecolare, come ad esempio GSH, per ridurre H2O2 e lipidi perossidi nei loro corrispondenti alcoli. Sono stati descritti quattro GSH-Pxs, codificati da diversi geni: GSH-Px-1 (cellulare GSH-Px) è onnipresente e riduce H2O2 e perossidi di acidi grassi, ma non lipidi perossilici esterificati.71 I lipidi esterificati vengono ridotti mediante GSH legato alla membrana -Px-4 (idroperossido fosfolipido GSH-Px), che può utilizzare diversi tioli a basso peso molecolare come equivalenti riducenti. GSH-Px-2 (gastrointestinale GSH-Px) è localizzato in cellule epiteliali gastrointestinali dove serve a ridurre i perossidi alimentari. 72 GSH-Px-3 (GSH-Px extracellulare) è l'unico membro della famiglia GSH-Px che risiede lo scomparto extracellulare e si ritiene che sia uno dei più importanti enzimi antiossidanti extracellulari nei mammiferi. Di questi, GSH-Px extracellulare è più ampiamente studiato nel polmone umano.73
Inoltre, lo smaltimento di H2O2 è strettamente associato a diversi enzimi contenenti tiolo, vale a dire TRX (TRX1 e TRX2), riduzione delle tioredossine (EC1.8.1.9) (TRRs), PRXs (che sono perossidasi tioredossina) e glutareossine.74
Due TRXs e TRRs sono stati caratterizzati in cellule umane, esistenti sia nel citosolo che nei mitocondri. Nel polmone, TRX e TRR sono espressi in epitelio bronchiale e alveolare e macrofagi. Sei differenti PRXs sono stati trovati nelle cellule umane, differente nella loro compartimentalizzazione ultrastrutturale. Studi sperimentali hanno rivelato l'importanza di PRX VI nella protezione dell'epitelio alveolare. Il polmone umano esprime tutti i PRX in epitelio bronchiale, epitelio alveolare e macrofagi. Recentemente è stato trovato che 75 PRX V funziona come una perossinitritica reduttasi, 76, il che significa che può funzionare come un potenziale composto protettivo nello sviluppo di lesioni polmonari ROS mediate .77
Comune a questi antiossidanti è il requisito di NADPH come equivalente riducente. NADPH mantiene catalasi nella forma attiva ed è utilizzato come cofattore da TRX e GSH reduttasi (EC 1.6.4.2), che converte GSSG in GSH, un co-substrato per gli GSH-Pxs. Il NADPH intracellulare, a sua volta, è generato dalla riduzione di NADP1 da glucose-6-fosfato deidrogenasi, il primo enzima e il tasso di limitazione del percorso di fosfato penato durante la conversione del glucosio-6-fosfato a 6-fosfogluconolattone. Con la generazione di NADPH, la deidrogenasi del glucosio-6-fosfato è un determinante critico della capacità di bufferizzazione GSH (GSH / GSSG) citosolica e, pertanto, può essere considerato un enzima antiossidante essenziale regolatore.78,79
I GST (EC 2.5.1.18), un'altra famiglia di enzimi antiossidanti, inattivano i metaboliti secondari, come le aldeidi insature, gli epossidi e gli idroperossidi. Sono state descritte tre famiglie principali di GST: GST citosolico, GST mitocondriale, 80,81 e GST microsomiale associato alla membrana che ha un ruolo nel metabolismo degli eicosanoidi e GSH.82 Sette classi di GST citosoliche sono identificate nei mammiferi, designati Alpha, Mu, Pi, Sigma, Theta, Omega e Zeta.83-86 Durante condizioni non stressate, i GST di classe Mu e Pi interagiscono con le chinasi Ask1 e JNK, rispettivamente, e inibiscono queste chinasi.87-89 È stato dimostrato che GSTP1 si dissocia JNK in risposta allo stress ossidativo.89 GSTP1 interagisce anche fisicamente con PRX VI e porta al recupero dell'attività dell'enzima PRX tramite glutationilazione della proteina ossidata.90
Antiossidanti non-enzimatici
Gli antiossidanti non-enzimatici comprendono composti a basso peso molecolare, come le vitamine (vitamine C e E), il carotene b, l'acido urico e GSH, un tripeptide (Lg-glutamil-L-cisteinil-L-glicina) che comprendono un tiolo sulfidril).
Vitamina C (Acido Ascorbico)
La vitamina C solubile in acqua (acido ascorbico) fornisce la capacità antiossidante acquosa a fase acquosa intracellulare e extracellulare principalmente mediante l'assorbimento di radicali liberi dell'ossigeno. Converte i radicali liberi della vitamina E verso la vitamina E. I suoi livelli plasmatici sono diminuiti con l'età.91,92
La vitamina E (a-tocopherolo)
La vitamina E solubile in lipidi è concentrata nel sito interno idrofobo della membrana cellulare e rappresenta la principale difesa contro il danneggiamento della membrana causato dagli ossidanti. La vitamina E dona l'elettrone al radicale perossilico, prodotta durante la perossidazione lipidica. a-Tocophero è la forma più attiva di vitamina E e il principale antiossidante legato alla membrana in cellule. La vitamina E innesca l'apoptosi delle cellule tumorali e inibisce le formazioni dei radicali liberi.93
Glutatione
GSH è altamente abbondante in tutti i compartimenti delle cellule ed è il principale antiossidante solubile. Il rapporto GSH / GSSG è un fattore determinante dello stress ossidativo. GSH mostra i suoi effetti antiossidanti in diversi modi. 94 Detossica perossido di idrogeno e perossidi lipidici tramite azione di GSH-Px. GSH dona il suo elettrone a H2O2 per ridurlo in H2O e O2. GSSG è nuovamente ridotto in GSH da GSH reduttasi che utilizza NAD (P) H come donatore di elettroni. GSH-Pxs sono anche importanti per la protezione della membrana cellulare dalla perossidazione dei lipidi. Il glutatione ridotto dona i protoni ai lipidi a membrana e li protegge dagli attacchi ossidanti.95
GSH è un cofattore per diversi enzimi disintossicante, come GSH-Px e transferasi. Ha un ruolo nella conversione della vitamina C ed E nelle loro forme attive. GSH protegge le cellule dall'apoptosi interagendo con percorsi di segnalazione proapoptotici e antiapoptotici. 94 Regola e attiva anche diversi fattori di trascrizione, quali AP-1, NF-kB e Sp-1.
Carotenoidi (b-Carotene)
I carotenoidi sono i pigmenti presenti nelle piante. In primo luogo, il carotenoide ha mostrato i suoi effetti antiossidanti in bassa pressione parziale dell'ossigeno, ma può avere effetti pro-ossidanti all'ossigeno più elevato. I carotenoidi perossilici (ROO), ossidrile (OH) e superossido (O22) concentrazioni. 96 Entrambi i carotenoidi e gli acidi retinoici (RAs) sono in grado di regolare i fattori di trascrizione.97 b-Carotene inibisce l'attivazione di NF-kB indotta dall'ossidante e l'interleuchina (IL) -98 e la produzione di fattore di necrosi tumorale. I carotenoidi influenzano anche l'apoptosi delle cellule. Gli effetti antiproliferativi della RA sono stati mostrati in diversi studi. Questo effetto di RA è mediato principalmente dai recettori dell'acido retinoico e varia tra i tipi di cellule. Nelle cellule del carcinoma mammario, il recettore dell'acido retinoico è stato dimostrato di inibire l'inibizione della crescita inducendo l'arresto del ciclo cellulare, l'apoptosi o entrambi. 6
L'EFFETTO DELLO STRESS OSSIDATIVO: MECCANISMI GENETICI, FISIOLOGICI E BIOCHIMICI
Lo stress ossidativo si verifica quando l'equilibrio tra antiossidanti e ROS viene interrotto a causa dell'esaurimento di antiossidanti o dell'accumulo di ROS. Quando si verifica lo sforzo ossidativo, le cellule cercano di contrastare gli effetti degli ossidanti e di ripristinare l'equilibrio redox mediante l'attivazione o la silenziazione di geni che codificano enzimi difensivi, fattori di transizione e proteine strutturali. Il rapporto tra glutatione ossidato e ridotto (101,102GSH / GSSG) è uno delle importanti determinanti dello stress ossidativo nel corpo. Una maggiore produzione di ROS nel corpo può cambiare la struttura del DNA, provocare la modifica delle proteine e dei lipidi, l'attivazione di diversi fattori di trascrizione indotti da stress e la produzione di citochine proinfiammatorie e antiinfiammatorie.
Effetti dello stress ossidativo sul DNA
ROS può portare a modifiche del DNA in diversi modi, che comportano il degrado delle basi, le interruzioni del DNA a singolo o doppio filamento, la purina, la pirimidina o le modifiche, le mutazioni, le delezioni o le traslocazioni legate al zucchero e la reticolazione con le proteine. La maggior parte di queste modificazioni del DNA (Fig. 1) sono altamente rilevanti per la carcinogenesi, l'invecchiamento e le malattie neurodegenerative, cardiovascolari e autoimmuni. Il fumo di tabacco, i metalli redox e i metalli non reattivi, come il ferro, il cadmio, il cromo e l'arsenico, sono anche coinvolti nella carcinogenesi e nell'invecchiamento generando radicali liberi o legandosi ai gruppi tiolici. La formazione di 8-OH-G è il più noto danno del DNA che si verifica con lo stress ossidativo e rappresenta un potenziale biomarker per la cancerogenesi.
Le regioni promotrici dei geni contengono sequenze di consenso per i fattori di trascrizione. Questi siti di legame del fattore di trascrizione contengono sequenze ricche di GC che sono suscettibili agli attacchi ossidanti. La formazione di 8-OH-G DNA nei siti di legame dei fattori di trascrizione può modificare il legame dei fattori di trascrizione e quindi cambiare l'espressione dei geni correlati come è stato dimostrato per le sequenze target AP-1 e Sp-1 Oltre a 103-OH-G, È stato anche dimostrato che 8 -ciclo-8,59 -deossiadenosina (ciclo-dA) inibisce la trascrizione da un gene reporter in un sistema cellulare se si trova in una scatola TATA.29 La proteina legante TATA avvia la trascrizione modificando la flessione del DNA . Il legame della proteina legante TATA può essere compromesso dalla presenza di cyclo-dA.
Lo stress ossidativo provoca l'instabilità di regioni di microsatellite (brevi tandem ripetizioni). Gli ioni di metallo attivo di Redox, i radicali idrossilici aumentano l'instabilità di microsatellite.105 Anche se le interruzioni di DNA a singolo filamento causate da lesioni all'ossidazione possono essere facilmente tollerate dalle cellule, le interruzioni del DNA a doppio filamento indotte da radiazioni ionizzanti possono essere una minaccia significativa per la sopravvivenza delle cellule.106
La metilazione nelle isole CpG del DNA è un importante meccanismo epigenetico che può provocare la silenziazione del gene. L'ossidazione di 5-MeCyt a 5-idrossimetil uracil (5-OHMeUra) può avvenire tramite reazioni di deinazione / ossidazione della timina o dei mesidi 5-idrossimetil citosina.107 Oltre all'espressione del gene modulante, anche la metilazione del DNA sembra influenzare l'organizzazione della cromatina.108 I modelli aberranti di metilazione del DNA indotti da attacchi ossidativi influenzano anche l'attività di riparazione del DNA.
Effetti dello stress ossidativo sui lipidi
I ROS possono indurre la perossidazione lipidica e interrompere la disposizione del doppio strato lipidico della membrana che può inattivare i recettori e gli enzimi legati alla membrana e aumentare la permeabilità dei tessuti.109 I prodotti della perossidazione lipidica, come l'MDA e le aldeidi insature, sono in grado di inattivare molte proteine cellulari formando la croce proteica -linkages.110-112 4-Hydroxy-2-nonenal provoca l'esaurimento del GSH intracellulare e induce la produzione di perossido, 113,114 attiva il recettore del fattore di crescita epidermico, 115 e induce la produzione di fibronectina.116 Prodotti della perossidazione lipidica, come isoprostani e sostanze reattive dell'acido tiobarbiturico , sono stati usati come biomarcatori indiretti dello stress ossidativo e sono stati mostrati livelli aumentati nel condensato del respiro esalato o nel liquido di lavaggio broncoalveolare o nel polmone di pazienti con malattia polmonare ostruttiva cronica o fumatori.
Effetti dello stress ossidativo sulle proteine
I ROS possono causare frammentazione della catena peptidica, alterazione della carica elettrica delle proteine, reticolazione delle proteine e ossidazione di amminoacidi specifici e quindi portare ad una maggiore suscettibilità alla proteolisi per degradazione da parte di proteasi specifiche.120 Cisteina e metionina residui nelle proteine sono particolarmente più suscettibile all'ossidazione.121 L'ossidazione dei gruppi sulfidrilici o dei residui di metionina delle proteine causa cambiamenti conformazionali, dispiegamento delle proteine e degradazione.8,121 123 Gli enzimi che hanno metalli sopra o vicino ai loro siti attivi sono particolarmente più sensibili all'ossidazione catalizzata dai metalli. È stato dimostrato che la modifica ossidativa degli enzimi inibisce le loro attività.124,125
In alcuni casi, può verificarsi un'ossidazione specifica delle proteine. Ad esempio, la metionina può essere ossidata di metionina sulfossido126 e fenilalanina a o-tirosina127; i gruppi di solfidril possono essere ossidati per formare legami di disolfuro; i gruppi 128 e carbonilici possono essere introdotti nelle catene laterali delle proteine. I raggi gamma, l'ossidazione catalizzata in metallo, l'HOCl e l'ozono possono causare la formazione di gruppi carbonilici.129
Effetti dello stress ossidativo sulla trasduzione del segnale
I ROS possono indurre l'espressione di diversi geni coinvolti nella trasduzione del segnale.1,130 Un rapporto elevato per GSH / GSSG è importante per la protezione della cellula dal danno ossidativo. L'interruzione di questo rapporto provoca l'attivazione di fattori di trascrizione redox sensibili, come NF-kB, AP-1, fattore nucleare delle cellule T attivate e fattore 1 inducibile dall'ipossia, che sono coinvolti nella risposta infiammatoria. L'attivazione dei fattori di trascrizione tramite ROS è ottenuta mediante cascate di trasduzione del segnale che trasmettono le informazioni dall'esterno all'interno della cellula. I recettori della tirosin chinasi, la maggior parte dei recettori del fattore di crescita, come il recettore del fattore di crescita epidermico, il recettore del fattore di crescita endoteliale vascolare e il recettore per il fattore di crescita derivato dalle piastrine, la proteina tirosina fosfatasi e la serina / treonina chinasi sono bersagli di ROS.131 Anche le chinasi extracellulari regolate dal segnale, JNK e p133, che sono membri della famiglia delle protein chinasi attivate da mitogeni e coinvolte in diversi processi cellulari, tra cui proliferazione, differenziazione e apoptosi, possono essere regolate da ossidanti.
In condizioni di stress ossidativo, i residui di cisteina nel sito di legame del DNA di c-Jun, alcune subunità AP-1 e la chinasi kB inibitoria subiscono una S-glutathiolation reversibile. È stato riportato che glutaredoxina e TRX svolgono un ruolo importante nella regolazione delle vie di segnalazione redox sensibili, come NF-kB e AP-1, proteina chinasi attivata da mitogeno p38 e JNK.134
NF-kB può essere attivato in risposta a condizioni di stress ossidativo, come ROS, radicali liberi e irradiazione UV.138 La fosforilazione di IkB libera NF-kB e gli consente di entrare nel nucleo per attivare la trascrizione genica.139 Un certo numero di chinasi ha è stato segnalato che fosforila IkB nei residui di serina. Queste chinasi sono i bersagli dei segnali ossidativi per l'attivazione di NF-kB.140 Gli agenti riducenti aumentano il legame del DNA di NF-kB, mentre gli agenti ossidanti inibiscono il legame del DNA di NF-kB. TRX può esercitare 2 azioni opposte nella regolazione di NF-kB: nel citoplasma, blocca la degradazione di IkB e inibisce l'attivazione di NF-kB ma migliora il legame del DNA di NF-kB nel nucleo.141 Attivazione di NF-kB tramite degradazione correlata all'ossidazione di IkB determina l'attivazione di diversi geni correlati alla difesa antiossidante. NF-kB regola l'espressione di diversi geni che partecipano alla risposta immunitaria, come IL-1b, IL-6, fattore di necrosi tumorale-a, IL-8 e diverse molecole di adesione.142,143 NF-kB regola anche l'angiogenesi e la proliferazione e differenziazione delle cellule.
AP-1 è anche regolato da stato redox. In presenza di H2O2, alcuni ioni metallici possono indurre l'attivazione di AP-1. L'aumento del rapporto di GSH / GSSG aumenta il legame AP-1 mentre GSSG inibisce il legame del DNA del DNA di AP-1.144 Il legame del DNA dell'eterodimero Fos / Jun è aumentato dalla riduzione di una singola cisteina conservata nel dominio di legame del DNA di ciascuno dei le proteine, 145 mentre il legame del DNA di AP-1 può essere inibito da GSSG in molti tipi di cellule, suggerendo che la formazione di legame di disolfuro dai residui di cisteina inibisce il legame del DNA di AP-1.146,147 La trasduzione del segnale attraverso lo stress ossidativo è riassunto in Figura 2.
CONCLUSIONI
Lo stress ossidativo può derivare dalla sovrapproduzione di ROS da reazioni metaboliche che utilizzano ossigeno e sposta l'equilibrio tra ossidante /antiossidante statistiche a favore degli ossidanti. ROS sono prodotti da attività metaboliche cellulari e fattori ambientali, come inquinanti atmosferici o fumo di sigaretta. I ROS sono molecole altamente reattive a causa di elettroni inattivi nella loro struttura e reagiscono con diverse macromolecole biologiche nella cellula, come i carboidrati, gli acidi nucleici, i lipidi e le proteine e alterano le loro funzioni. ROS colpisce anche l'espressione di diversi geni mediante l'upregulation di fattori di trascrizione sensibili alla redox e il rimodellamento della cromatina mediante alterazione dell'acetilazione / deacetilazione dell'istone. La regolazione dello stato redox è fondamentale per la vitalità delle cellule, l'attivazione, la proliferazione e la funzione dell'organo.
BIBLIOGRAFIA
1. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Radicali liberi, metalli e antiossidanti nel cancro indotto da stress ossidativo. Chem Biol Interact. 2006; 160: 1 40.
2. Halliwell B, Gutteridge JMC. Radici liberi in biologia e medicina. 3rd ed. New York: Oxford University Press; 1999.
3. Marnett LJ. Perossidazione lipidica Danno del DNA da parte della malondialdeide. Mutat Res. 1999; 424: 83.
4. Siems WG, Grune T, Esterbauer H. 4-Hydroxynonenal formazione durante ischemia e riperfusione dell'intestino tenue di ratto. Life Sci. 1995; 57: 785-789.
5. Stadtman ER. Ruolo delle specie ossidanti nell'invecchiamento. Curr Med Chem. 2004; 11: 1105-1112.
6. Wang MY, Dhingra K, Hittelman WN, Liehr JG, deAndrade M, Li DH. Malondialdeide putativa indotta dalla perossidazione lipidica Addotti del DNA nei tessuti del seno umano. Biomarcatori dell'epidemiolo del cancro Prev. 1996; 5: 705 710.
7. Jenner P. Stress ossidativo nella malattia di Parkinson. Ann Neurol. 2003; 53: S26 S36.
8. Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. Una valutazione del danno ossidativo a proteine, lipidi e DNA nel cervello da pazienti con malattia di Alzheimer. J Neurochem. 1997; 68: 2061-2069.
9. Sayre LM, Smith MA, Perry G. Chimica e biochimica dello stress ossidativo nelle malattie neurodegenerative. Curr Med Chem. 2001; 8: 721 738.
10. Toshniwal PK, Zarling EJ. Prove per un aumento della perossidazione lipidica nella sclerosi multipla. Neurochem Res. 1992; 17: 205-207.
11. Dhalla NS, Temsah RM, Netticadan T. Ruolo dello stress ossidativo nelle malattie cardiovascolari. J Hypertens. 2000; 18: 655 673.
12. Kasparova S, Brezova V, Valko M, Horecky J, Mlynarik V, et al. Studio dello stress ossidativo in un modello di ratto di ipoperfusione cerebrale cronica. Neurochem Int. 2005; 46: 601 611.
13. Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. La produzione di anioni superossido è aumentata in un modello di ipertensione genetica: ruolo dell'endotelio. Ipertensione. 1999; 33: 1353 1358.
14. Kukreja RC, Hess ML. Il sistema dei radicali liberi dell'ossigeno: dalle equazioni attraverso le interazioni delle proteine di membrana al danno e protezione cardiovascolare Cardiovasc Res. 1992; 26: 641 655.
15. Asami S, Manabe H, Miyake J, Tsurudome Y, Hirano T, et al. Il fumo di sigaretta induce un aumento del danno ossidativo al DNA, 8-idrossideossiguanosina, in un sito centrale del polmone umano. Cancerogenesi. 1997; 18: 1763-1766.
16. Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Eventi ossidativi e nitrosativi nell'asma. Radic Biol Med. 2003; 35: 213-225.
17. Comhair SA, Ricci KS, Arroliga M, Lara AR, Dweik RA, et al. Correlazione del deficit sistemico di superossido dismutasi con l'ostruzione del flusso d'aria nell'asma. Sono J Respir Crit Care Med. 2005; 172: 306-313.
18. Comhair SA, Xu W, Ghosh S, Thunnissen FB, Almasan A, et al. Inattivazione della superossido dismutasi nella fisiopatologia del rimodellamento e della reattività delle vie aeree asmatiche. Sono J Pathol. 2005; 166: 663 674.
19. Dut R, Dizdar EA, Birben E, Sackesen C, Soyer OU, Besler T, Kalayci O. Stress ossidativo e suoi determinanti nelle vie aeree dei bambini con asma. Allergia. 2008; 63: 1605-1609.
20. Ercan H, Birben E, Dizdar EA, Keskin O, Karaaslan C, et al. Stress ossidativo e determinanti genetici ed epidemiologici del danno ossidativo nell'asma infantile. J Allergy Clin Immunol. 2006; 118: 1097-1104.
21. Fitzpatrick AM, Teague WG, Holguin F, Yeh M, Brown LA. Programma di ricerca sull'asma grave. L'omeostasi del glutatione delle vie aeree è alterata nei bambini con asma grave: evidenza di stress ossidante. J Allergy Clin Immunol. 2009; 123: 146.
22. Miller DM, Buettner GR, Aust SD. Metalli di transizione come catalizzatori di reazioni di “autoossidazione”. Radic Biol Med. 1990; 8: 95-108.
23. Dupuy C, Virion A, Ohayon R, Kaniewski J, Déme D, Pommier J. Meccanismo di formazione del perossido di idrogeno catalizzato dalla NADPH ossidasi nella membrana plasmatica tiroidea. J Biol Chem. 1991; 266: 3739 3743.
24. Granger DN. Ruolo della xantina ossidasi e dei granulociti nel danno da ischemiareperfusione. Sono J Physiol. 1988; 255: H1269 H1275.
25. Fenton HJH. Ossidazione dell'acido tartarico in presenza di ferro. J Chem Soc. 1984; 65: 899-910.
26. Haber F, Weiss JJ. La decomposizione catalitica del perossido di idrogeno da parte dei sali di ferro. Proc R Soc Lond Ser A. 1934; 147: 332.
27. Liochev SI, Fridovich I. L'Haber Weiss percorse 70 anni dopo: una visione alternativa. Redox Rep.2002; 7: 55.
28. Klebanoff SJ. Mieloperossidasi: amico e nemico. J Leukoc Biol. 2005; 77: 598 625.
29. Whiteman M, Jenner A, Halliwell B. Modificazioni di base indotte da acido ipocloroso nel DNA del timo di vitello isolato. Chem Res Toxicol. 1997; 10: 1240-1246.
30. Kulcharyk PA, Heinecke JW. L'acido ipocloroso prodotto dal sistema mieloperossidasi dei fagociti umani induce legami incrociati covalenti tra DNA e proteine. Biochimica. 2001; 40: 3648.
31. Brennan ML, Wu W, Fu X, Shen Z, Song W, et al. Un racconto di due controversie: definire sia il ruolo delle perossidasi nella formazione di nitrotirosina in vivo utilizzando perossidasi eosinofila e topi carenti di mieloperossidasi, sia la natura delle specie di azoto reattivo generate dalla perossidasi. J Biol Chem. 2002; 277: 17415 17427.
32. Denzler KL, Borchers MT, Crosby JR, Cieslewicz G, Hines EM, et al. La degranulazione degli eosinofili e l'ossidazione mediata dalla perossidasi delle proteine delle vie aeree non si verificano in un modello di infiammazione polmonare provocato dall'ovalbumina di topo. J Immunol. 2001; 167: 1672-1682.
33. van Dalen CJ, Winterbourn CC, Senthilmohan R, Kettle AJ. Nitrito come substrato e inibitore della mieloperossidasi. Implicazioni per la nitrazione e la produzione di acido ipocloroso nei siti di infiammazione. J Biol Chem. 2000; 275: 11638 11644.
34. Legno LG, Fitzgerald DA, Gibson PG, Cooper DM, Garg ML. La perossidazione lipidica determinata dagli isoprostani plasmatici è correlata alla gravità della malattia nell'asma lieve. Lipidi. 2000; 35: 967 974.
35. Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Kharitonov SA, Barnes PJ. 8-isoprostano aumentato, un marker di stress ossidativo, nel condensato esalato dei pazienti asmatici. Sono J Respir Crit Care Med. 1999; 160: 216-220.
36. Chiesa DF, Pryor WA. Chimica dei radicali liberi del fumo di sigaretta e sue implicazioni tossicologiche. Prospettiva della salute ambientale. 1985; 64: 111 126.
37. Hiltermann JT, Lapperre TS, van Bree L, Steerenberg PA, Brahim JJ, et al. L'infiammazione indotta dall'ozono valutata nell'espettorato e nel liquido di lavaggio bronchiale da asmatici: un nuovo strumento non invasivo negli studi epidemiologici sull'inquinamento atmosferico e l'asma. Radic Biol Med. 1999; 27: 1448-1454.
38. Nightingale JA, Rogers DF, Barnes PJ. Effetto dell'ozono inalato sull'ossido nitrico esalato, sulla funzione polmonare e sull'espettorato indotto in soggetti normali e asmatici. Torace. 1999; 54: 1061-1069.
39. Cho AK, Sioutas C, Miguel AH, Kumagai Y, Schmitz DA, et al. Attività redox del particolato aerodisperso in diversi siti nel bacino di Los Angeles. Environ Res. 2005; 99: 40.
40. Comhair SA, Thomassen MJ, Erzurum SC. Induzione differenziale della glutatione perossidasi extracellulare e dell'ossido nitrico sintasi 2 nelle vie aeree di individui sani esposti al 100% di O (2) o al fumo di sigaretta. Am J Respir Cell Mol Biol. 2000; 23: 350 354.
41. Matthay MA, Geiser T, Matalon S, Ischiropoulos H. Lesione polmonare mediata dall'ossidante nella sindrome da distress respiratorio acuto. Crit Care Med. 1999; 27: 2028-2030.
42. Biaglow JE, Mitchell JB, Held K. L'importanza del perossido e del superossido nella risposta ai raggi X. Int J Radiat Oncol Biol Phys. 1992; 22: 665.
43. Chiu SM, Xue LY, Friedman LR, Oleinick NL. Sensibilizzazione mediata da ioni di rame dei siti di attacco della matrice nucleare alle radiazioni ionizzanti. Biochimica. 1993; 32: 6214 6219.
44. Narayanan PK, Goodwin EH, Lehnert BE. Le particelle alfa avviano la produzione biologica di anioni superossido e perossido di idrogeno nelle cellule umane. Cancer Res. 1997; 57: 3963 3971.
45. Tuttle SW, Varnes ME, Mitchell JB, Biaglow JE. Sensibilità agli ossidanti chimici e alle radiazioni nelle linee cellulari CHO carenti nell'attività del ciclo pentoso ossidativo. Int J Radiat Oncol Biol Phys. 1992; 22: 671 675.
46. Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, et al. Manganese
espressione genica mediata dalla superossido dismutasi in radiazioni indotte
risposte adattive. Mol Cell Biol. 2003; 23: 2362-2378.
47. Azzam EI, de Toledo SM, Spitz DR, Little JB. Metabolismo ossidativo
modulano la trasduzione del segnale e la formazione di micronucleo in soggetti esterni
cellule di fibroblasti normali umano irradiati da una particella. Cancer Res.
2002; 62: 5436 5442.
48. Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB.
Generazione ionizzante-inducibile, mitocondri-dipendente di reattivo
ossigeno / azoto. Cancer Res. 2001; 61: 3894-3901.
49. Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK percorsi in
risposte alle radiazioni. Oncogene. 2003; 22: 5885 5896.
50. Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, et al. tioredossina
la traslocazione nucleare e l'interazione con il fattore redox-1 attiva il fattore di trascrizione AP-1 in risposta alle radiazioni ionizzanti. Cancer Res. 2000; 60: 6688 6695.
51. Cadet J, Douki T, Gasparutto D, Ravanat JL. Danno ossidativo al DNA: formazione, misurazione e caratteristiche biochimiche. Mutat Res. 2003; 531: 5 23.
52. Yokoya A, Cunniffe SM, O'Neill P. Effetto dell'idratazione sull'induzione di rotture del filamento e lesioni alla base nei film di DNA plasmidico mediante gammaradiazione. J Am Chem Soc. 2002; 124: 8859 8866.
53. Janssen YM, Van Houten B, Borm PJ, Mossman BT. Risposte di cellule e tessuti al danno ossidativo. Lab Invest. 1993; 69: 261-274.
54. Iwanaga M, Mori K, Iida T, Urata Y, Matsuo T, et al. Induzione dipendente dal fattore nucleare kappa B della gamma glutamilcisteina sintetasi mediante radiazioni ionizzanti in cellule di glioblastoma umano T98G. Radic Biol Med. 1998; 24: 1256 1268.
55. Stohs SJ, Bagchi D. Meccanismi ossidativi nella tossicità degli ioni metallici. Radic Biol Med. 1995; 18: 321 336.
56. Leonard SS, Harris GK, Shi X. Stress ossidativo indotto da metalli e trasduzione del segnale. Radic Biol Med. 2004; 37: 1921-1942.
57. Shi H, Shi X, Liu KJ. Meccanismo ossidativo della tossicità da arsenico e cancerogenesi. Mol Cell Biochem. 2004; 255: 67.
58. Pi J, Horiguchi S, Sun Y, Nikaido M, Shimojo N, Hayashi T. Un potenziale meccanismo per la compromissione della formazione di ossido nitrico causata da una prolungata esposizione orale all'arseniato nei conigli. Gratuito Radic Biol Med.2003; 35: 102 113.
59. Rin K, Kawaguchi K, Yamanaka K, Tezuka M, Oku N, Okada S. Le rotture di DNAstrand indotte dall'acido dimetilarsinico, un metabolita degli arsenici inorganici, sono fortemente potenziate dai radicali anionici superossido. Biol Pharm Bull. 1995; 18: 45.
60. Waalkes MP, Liu J, Ward JM, Diwan LA. Meccanismi alla base della carcinogenesi dell'arsenico: ipersensibilità dei topi esposti all'arsenico inorganico durante la gestazione. Tossicologia. 2004; 198: 31.
61. Schiller CM, Fowler BA, Woods JS. Effetti dell'arsenico sull'attivazione della piruvato deidrogenasi. Prospettiva della salute ambientale. 1977; 19: 205-207.
62. Monterio HP, Bechara EJH, Abdalla DSP. Coinvolgimento dei radicali liberi nelle porfirie neurologiche e avvelenamento da piombo. Mol Cell Biochem. 1991; 103: 73.
63. Tripathi RM, Raghunath R, Mahapatra S. Piombo nel sangue e il suo effetto sui livelli di Cd, Cu, Zn, Fe e di emoglobina dei bambini. Sci Total Environ. 2001; 277: 161.
64. Nehru B, Dua R. L'effetto del selenio nella dieta sulla neurotossicità del piombo. J Environ Pathol Toxicol Oncol. 1997; 16: 47.
65. Reid TM, Feig DI, Loeb LA. Mutagenesi da radicali dell'ossigeno indotti dai metalli. Prospettiva della salute ambientale. 1994; 102 (suppl 3): 57.
66. Kinnula VL, Crapo JD. Superossido dismutasi nel polmone e malattie polmonari umane. Sono J Respir Crit Care Med. 2003; 167: 1600-1619.
67. Kinnula VL. Produzione e degradazione dei metaboliti dell'ossigeno durante gli stati infiammatori nel polmone umano. La droga di Curr prende di mira l'allergia alle infiammazioni. 2005; 4: 465-470.
68. Zelko IN, Mariani TJ, Folz RJ. Famiglia multigenica di superossido dismutasi: un confronto tra le strutture, l'evoluzione e l'espressione del gene CuZn-SOD (SOD1), Mn-SOD (SOD2) ed EC-SOD (SOD3). Radic Biol Med. 2002; 33: 337.
69. Kirkman HN, Rolfo M, Ferraris AM, Gaetani GF. Meccanismi di protezione della catalasi da NADPH. Cinetica e stechiometria. J Biol Chem. 1999; 274: 13908 13914.
70. Floh L. Glutatione perossidasi. Basic Life Sci. 1988; 49: 663-668.
71. Arthur JR. Il glutatione perossidasi. Cell Mol Life Sci. 2000; 57: 1825-1835.
72. Chu FF, Doroshow JH, Esworthy RS. Espressione, caratterizzazione e distribuzione tissutale di una nuova glutatione perossidasi dipendente dal selenio cellulare, GSHPx-GI. J Biol Chem. 1993; 268: 2571 2576.
73. Comhair SA, Bhathena PR, Farver C, Thunnissen FB, Erzurum SC. Induzione extracellulare della glutatione perossidasi nei polmoni asmatici: prove per la regolazione redox dell'espressione nelle cellule epiteliali delle vie aeree umane. FASEB J. 2001; 15: 70-78.
74. Gromer S, Urig S, Becker K. Il sistema tioredossinico dalla scienza alla clinica. Med Res Rev.2004; 24: 40 89.
75. Kinnula VL, Lehtonen S, Kaarteenaho-Wiik R, Lakari E, P kk P, et al. Espressione cellulare specifica delle perossirossine nella sarcoidosi polmonare e polmonare umana. Torace. 2002; 57: 157.
76. Dubuisson M, Vander Stricht D, Clippe A, Etienne F, Nauser T, et al. La perossirossina 5 umana è una perossinitrito reduttasi. FEBS Lett. 2004; 571: 161.
77. Holmgren A. Funzione antiossidante dei sistemi tioredossina e glutaredossina. Segnale Redox antiossidante. 2000; 2: 811 820.
78. Dickinson DA, Forman HJ. Glutatione in difesa e segnalazione: lezioni da un piccolo tiolo. Ann NY Acad Sci. 2002; 973: 488 504.
79. Sies H. glutatione e suo ruolo nelle funzioni cellulari. Radic Biol Med. 1999; 27: 916-921.
80. Ladner JE, Parsons JF, Rife CL, Gilliland GL, Armstrong RN. Vie evolutive parallele per glutatione transferasi: struttura e meccanismo dell'enzima kappa di classe mitocondriale rGSTK1-1. Biochimica. 2004; 43: 52.
81. Robinson A, Huttley GA, Booth HS, Board PG. Studi di modellazione e bioinformatica della glutatione transferasi di classe kappa umana prevedono una nuova famiglia di transferasi terza con omologia alle isomerasi procariotiche 2-idrossocromene-2-carbossilato. Biochem J. 2004; 379: 541.
82. Jakobsson PJ, Morgenstern R, Mancini J, Ford-Hutchinson A, Persson B. Caratteristiche strutturali comuni della superfamiglia diffusa MAPEGda di proteine associate alla membrana con funzioni altamente divergenti nel metabolismo degli eicosanoidi e del glutatione. Protein Sci. 1999; 8: 689 692.
83. Hayes JD, Pulford DJ. La famiglia dei supergeni glutatione S-transferasi: regolazione della GST e contributo degli isoenzimi alla chemioprotezione del cancro e alla resistenza ai farmaci. Crit Rev Biochem Mol Biol. 1995; 30: 445.
84. Armstrong RN. Struttura, meccanismo catalitico ed evoluzione delle transferasi del glutatione. Chem Res Toxicol. 1997; 10: 2 18.
85. Hayes JD, McLellan LI. Il glutatione e gli enzimi dipendenti dal glutatione rappresentano una difesa coordinata contro lo stress ossidativo. Radic Res. 1999; 31: 273 300.
86. Sheehan D, Meade G, Foley VM, Dowd CA. Struttura, funzione ed evoluzione delle transferasi del glutatione: implicazioni per la classificazione dei membri non mammiferi di un'antica superfamiglia enzimatica. Biochem J. 2001; 360: 1 16.
87. Cho SG, Lee YH, Park HS, Ryoo K, Kang KW, et al. La glutatione S-transferasi Mu modula i segnali attivati dallo stress sopprimendo la chinasi di regolazione del segnale di apoptosi 1. J Biol Chem. 2001; 276: 12749 12755.
88. Dorion S, Lambert H, Landry J. L'attivazione della via di segnalazione p38 da shock termico coinvolge la dissociazione della glutatione S-transferasi Mu da Ask1. J Biol Chem. 2002; 277: 30792-30797.
89. Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, et al. Regolazione della segnalazione JNK da parte di GSTp. EMBO J. 1999; 18: 1321.
90. Manevich Y, Feinstein SI, Fisher AB. L'attivazione dell'enzima antiossidante 1-CYS perossiredossina richiede la glutationilazione mediata dall'eterodimerizzazione con pGST. Proc Natl Acad Sci US A. 2004; 101: 3780-3785.
91. Bunker VW. Radicali liberi, antiossidanti e invecchiamento. Med Lab Sci. 1992; 49: 299-312.
92. Mezzetti A, Lapenna D, Romano F, Costantini F, Pierdomenico SD, et al. Stress ossidativo sistemico e sua relazione con l'età e la malattia. J Am Geriatr Soc. 1996; 44: 823 827.
93. White E, Shannon JS, Patterson RE. Rapporto tra vitamina e
uso di integratori di calcio e cancro al colon. Biomarcatori dell'epidemiolo del cancro Prev. 1997; 6: 769-774.
94. Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C. Nuovi meccanismi di composti antiossidanti naturali nei sistemi biologici: coinvolgimento del glutatione e degli enzimi correlati al glutatione. J Nutr Biochem. 2005; 16: 577 586.
95. Curello S, Ceconi C, Bigoli C, Ferrari R, Albertini A, Guarnieri C. Cambiamenti nello stato del glutatione cardiaco dopo ischemia e riperfusione. Experientia. 1985; 41: 42 43.
96. El-Agamey A, Lowe GM, McGarvey DJ, Mortensen A, Phillip DM, Truscott TG. Chimica dei radicali carotenoidi e proprietà antiossidanti / proossidanti. Arch Biochem Biophys. 2004; 430: 37 48.
97. Rice-Evans CA, Sampson J, Bramley PM, Holloway DE. Perché ci aspettiamo che i carotenoidi siano antiossidanti in vivo? Radic Res. 1997; 26: 381.
98. Niles RM. Vie di segnalazione nella chemioprevenzione dei retinoidi e nel trattamento del cancro. Mutat Res. 2004; 555: 81 96.
99. Donato LJ, Noy N. Soppressione della crescita del carcinoma mammario da parte dell'acido retinoico: i geni proapoptotici sono bersagli per il recettore dell'acido retinoico e la segnalazione della proteina II legante l'acido retinoico cellulare. Cancer Res. 2005; 65: 8193 8199.
100. Niizuma H, Nakamura Y, Ozaki T, Nakanishi H, Ohira M, et al. Bcl-2 è un regolatore chiave per la morte cellulare apoptotica indotta dall'acido retinoico nel neuroblastoma. Oncogene. 2006; 25: 5046 5055.
101. Dalton TP, Shertzer HG, Puga A. Regolazione dell'espressione genica da ossigeno reattivo. Ann Rev Pharmacol Toxicol. 1999; 39: 67-101.
102. Scandalios JG. Risposte genomiche allo stress ossidativo. In: Meyers RA, ed. Enciclopedia di biologia cellulare molecolare e medicina molecolare. Vol 5. 2a ed. Weinheim, Germania: Wiley-VCH; 2004: 489-512.
103. Ghosh R, Mitchell DL. Effetto del danno ossidativo al DNA negli elementi promotori sul legame del fattore di trascrizione. Ris. Acidi nucleici 1999; 27: 3213 3218.
104. Marietta C, Gulam H, Brooks PJ. Una singola lesione di 8, 50-ciclo-20-deossiadenosina in una scatola TATA impedisce il legame della proteina legante TATA e riduce fortemente la trascrizione in vivo. Riparazione del DNA (Amst). 2002; 1: 967-975.
105. Jackson AL, Chen R, Loeb LA. Induzione dell'instabilità microsatellitica
da danno ossidativo al DNA. Proc Natl Acad Sci US A. 1998; 95: 12468 12473.
106. Caldecott KW. Interazioni proteina-proteina durante la riparazione della rottura di un singolo filamento del DNA dei mammiferi. Biochem Soc Trans. 2003; 31: 247 251.
107. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Danno ossidativo al DNA: meccanismi, mutazione e malattia. FASEB J. 2003; 17: 1195-1214.
108. Jones PL, Wolffe AP. Relazioni tra organizzazione della cromatina e metilazione del DNA nella determinazione dell'espressione genica. Semin Cancer Biol. 1999; 9: 339.
109. Girotti AW. Meccanismi di perossidazione lipidica. J Free Radic Biol Med. 1985; 1: 87.
110. Siu GM, Draper HH. Metabolismo della malonaldeide in vivo e in vitro. Lipidi. 1982; 17: 349 355.
111. Esterbauer H, Koller E, Slee RG, Koster JF. Possibile coinvolgimento del prodotto di perossidazione lipidica 4-idrossinonenale nella formazione di cromolipidi fluorescenti. Biochem J. 1986; 239: 405 409.
112. Hagihara M, Nishigaki I, Maseki M, Yagi K. Cambiamenti dipendenti dall'età nei livelli di perossido lipidico nelle frazioni lipoproteiche del siero umano. J Gerontol. 1984; 39: 269-272.
113. Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, et al. 4- Hydroxynonenal, un prodotto aldeidico della perossidazione lipidica di membrana, altera il trasporto del glutammato e la funzione mitocondriale nei sinaptosomi. Neuroscienza. 1997; 806: 85.
114. Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Attivazione delle vie di segnalazione dello stress dal prodotto finale della perossidazione lipidica. Il 4-idrossi-2-nonenale è un potenziale induttore della produzione di perossido intracellulare. J Biol Chem. 1999; 274: 2234-2242.
115. Suc I, Meilhac O, Lajoie-Mazenc I, Vandaele J, Jurgens G, Salvayre R, Negre-Salvayre A. Attivazione del recettore EGF da LDL ossidato. FASEB J. 1998; 12: 665.
116. Tsukagoshi H, Kawata T, Shimizu Y, Ishizuka T, Dobashi K, Mori M. 4-Hydroxy-2-nonenal migliora la produzione di fibronectina da parte dei fibroblasti polmonari umani IMR-90 in parte tramite l'attivazione del segnale extracellulare legato al recettore del fattore di crescita epidermico- via regolata della chinasi p44 / 42. Toxicol Appl Pharmacol. 2002; 184: 127 135.
117. Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ. 8-isoprostano esalato come biomarcatore in vivo dello stress ossidativo polmonare in pazienti con BPCO e fumatori sani. Sono J Respir Crit Care Med. 2000; 162: 1175-1177.
118. Morrison D, Rahman I, Lannan S, MacNee W. Permeabilità epiteliale, infiammazione e stress ossidante negli spazi aerei dei fumatori. Sono J Respir Crit Care Med. 1999; 159: 473.
119. Nowak D, Kasielski M, Antczak A, Pietras T, Bialasiewicz P. Aumento del contenuto di sostanze reattive all'acido tiobarbiturico e perossido di idrogeno nel condensato del respiro espirato di pazienti con malattia polmonare ostruttiva cronica stabile: nessun effetto significativo del fumo di sigaretta. Respir Med. 1999; 93: 389.
120. Kelly FJ, Mudway IS. Ossidazione delle proteine all'interfaccia aria-polmone. Aminoacidi. 2003; 25: 375 396.
121. Dean RT, Roberts CR, Jessup W. Frammentazione di polipeptidi extracellulari e intracellulari da radicali liberi. Prog Clin Biol Res. 1985; 180: 341-350.
122. Keck RG. L'uso del t-butil idroperossido come sonda per l'ossidazione della metionina nelle proteine. Biochimica anale. 1996; 236: 56.
123. Davies KJ. Danni alle proteine e degradazione da parte dei radicali dell'ossigeno. I. Aspetti generali. J Biol Chem. 1987; 262: 9895 9901.
124. Stadtman ER. L'ossidazione ioni catalizzata metallica delle proteine: meccanismo biochimico e conseguenze biologiche. Libero Radic Biol Med.
1990; 9: 315 325.
125. Fucci L, Oliver CN, Coon MJ, Stadtman ER. Inattivazione di enzimi metabolici chiave da reazioni di ossidazione a funzione mista: possibile implicazione nel turnover proteico e nell'invecchiamento. Proc Natl Acad Sci US A. 1983; 80: 1521-1525.
126. Stadtman ER, Moskovitz J, Levine RL. Ossidazione dei residui di metionina delle proteine: conseguenze biologiche. Segnale Redox antiossidante. 2003; 5: 577 582.
127. Stadtman ER, Levine RL. Ossidazione mediata da radicali liberi di amminoacidi liberi e residui amminoacidici nelle proteine. Aminoacidi. 2003; 25: 207-218.
128. Stadtman ER. Ossidazione delle proteine nell'invecchiamento e nelle malattie legate all'età. Ann NY Acad Sci. 2001; 928: 22.
129. Shacter E. Quantificazione e significato dell'ossidazione delle proteine in campioni biologici. Drug Metab Rev.2000; 32: 307 326.
130. Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Stress ossidativo e segnalazione cellulare. Curr Med Chem. 2004; 11: 1163-1182.
131. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Fattore di crescita endoteliale vascolare (VEGF) e suoi recettori. FASEB J. 1999; 13: 9-22.
132. Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, et al. Regolazione della generazione di specie reattive dell'ossigeno nei fibroblasti mediante Rac1. Biochem J. 1996; 318: 379.
133. Sun T, Oberley LW. Regolazione redox degli attivatori trascrizionali. Radic Biol Med. 1996; 21: 335-348.
134. Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Redox regolazione del legame c-Jun DNA mediante S-glutatiolazione reversibile. FASEB J. 1999; 13: 1481-1490.
135. Reynaert NL, Ckless K, Guala AS, Wouters EF, van der Vliet A, Janssen Heininger
YM. Rilevazione in situ di proteine S-glutationilate dopo derivatizzazione di cisteina catalizzata da glutaredossina-1. Biochim Biophys Acta. 2006; 1760: 380.
136. Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulazione di glutaredoxina-1
espressione in un modello murino di malattia allergica delle vie aeree. Am J Respir Cell Mol Biol. 2007; 36: 147.
137. Filomeni G, Rotilio G, Ciriolo MR. Segnalazione cellulare e sistema redox del glutatione. Biochem Pharmacol. 2002; 64: 1057-1064.
138. Pande V, Ramos MJ. Riconoscimento molecolare della prostaglandina J (15) 12,14-deoxydelta (2) da parte del fattore nucleare-kappa B e di altre proteine cellulari. Bioorg Med Chem Lett. 2005; 15: 4057 4063.
139. Perkins ND. Integrazione delle vie di segnalazione cellulare con la funzione NF-kappaB e IKK. Nat Rev Mol Cell Biol. 2007; 8: 49.
140. Gilmore TD. Introduzione a NF-kappaB: attori, percorsi, prospettive. Oncogene. 2006; 25: 6680 6684.
141. Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Ruoli distinti della tioredossina nel citoplasma e nel nucleo. Un meccanismo a due fasi di regolazione redox del fattore di trascrizione NF-kappaB. J Biol Chem. 1999; 274: 27891 27897.
142. Ward PA. Ruolo del complemento, delle chemochine e delle citochine regolatrici nel danno polmonare acuto. Ann NY Acad Sci. 1996; 796: 104-112.
143. Akira S, Kishimoto A. NF-IL6 e NF-kB nella regolazione genica delle citochine. Adv Immunol. 1997; 65: 1 46.
144. Meyer M, Schreck R, Baeuerle PA. L'H2O2 e gli antiossidanti hanno effetti opposti sull'attivazione di NF-kappa B e AP-1 nelle cellule intatte: AP-1 come fattore di risposta antiossidante secondario. EMBO J. 1993; 12: 2005-2015.
145. Abate C, Patel L, Rausher FJ, Curran T. Redox regolazione di fos e Jun DNA-binding attività in vitro. Scienza. 1990; 249: 1157-1161.
146. Galter D, Mihm S, Droge W. Effetti distinti del glutatione disolfuro sui fattori di trascrizione nucleare kB e sulla proteina attivatrice-1. Eur J Biochem. 1994; 221: 639.
147. L'attività trascrizionale di Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 è regolata da un'associazione diretta tra tioredossina e Ref-1. Proc Natl Acad Sci US A. 1997; 94: 3633-3638.
Lo strumento Find A Practitioner di IFM è la più grande rete di riferimento in Medicina Funzionale, creata per aiutare i pazienti a localizzare professionisti di Medicina Funzionale in qualsiasi parte del mondo. I professionisti certificati IFM sono elencati per primi nei risultati di ricerca, data la loro vasta formazione in Medicina Funzionale
Prenotazione online e appuntamenti 24 ore su 7, XNUMX giorni su XNUMX *